

Lamin A/C-mediated neuromuscular junction defects in Emery-Dreifuss muscular dystrophy
- PMID: 19124654
- PMCID: PMC2615092
- DOI: 10.1083/jcb.200811035
Lamin A/C-mediated neuromuscular junction defects in Emery-Dreifuss muscular dystrophy
Abstract
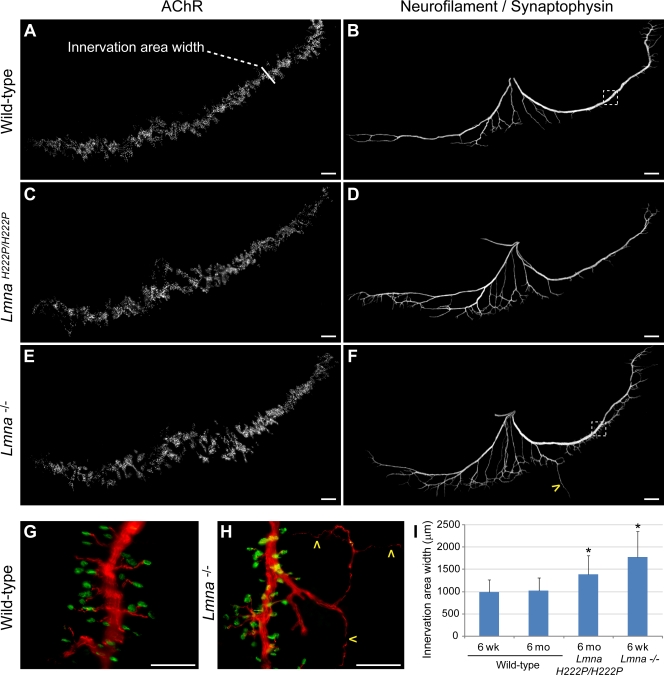
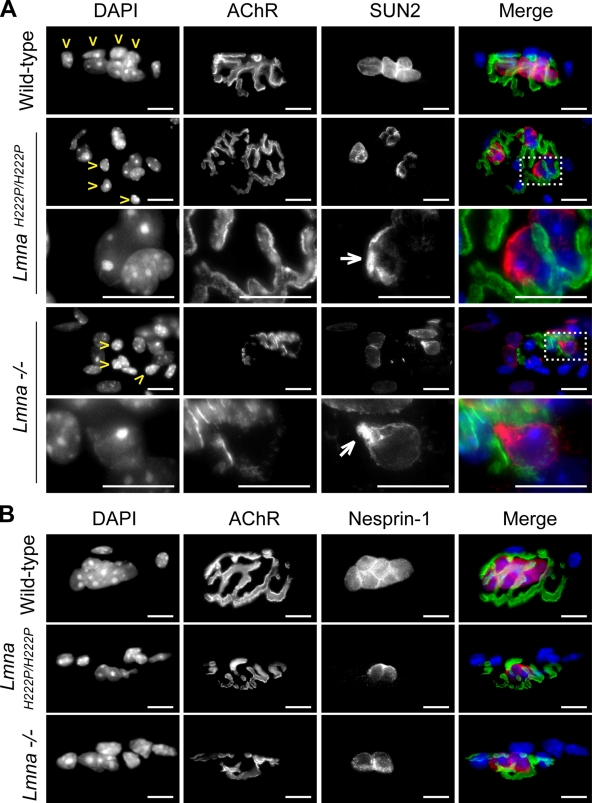
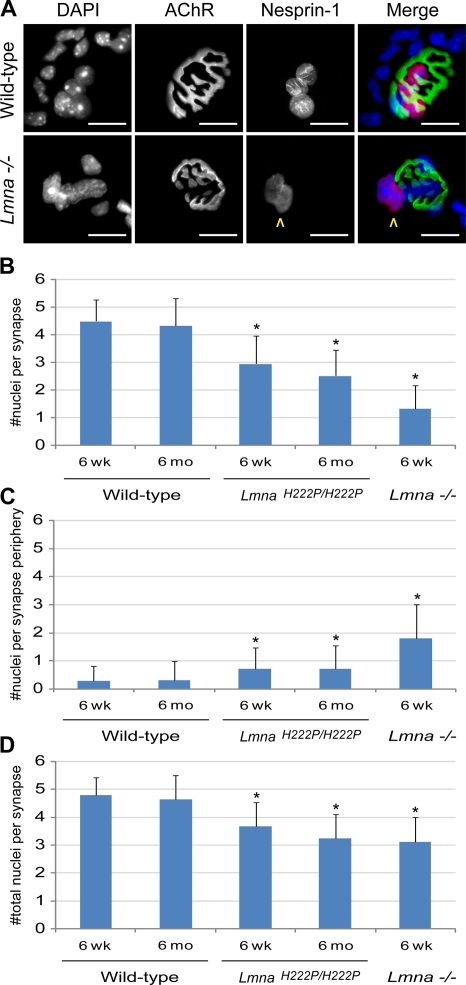
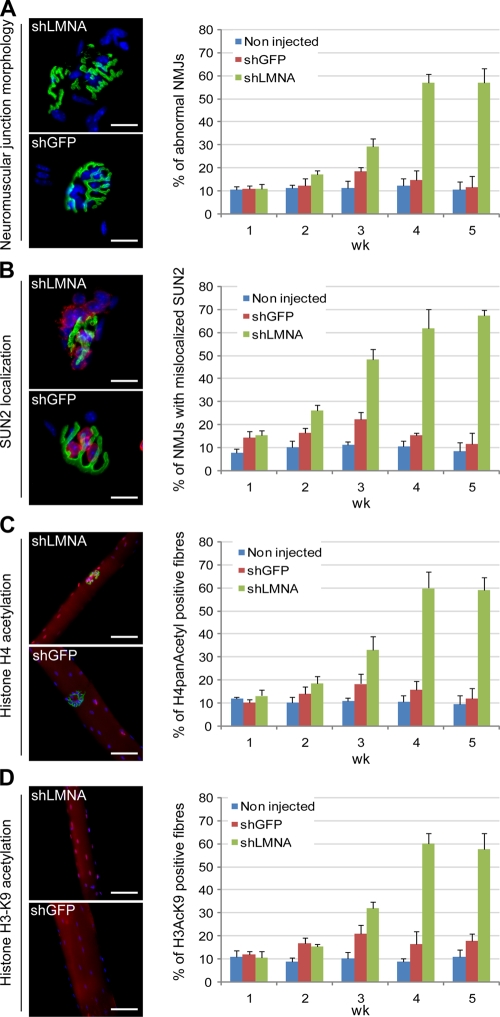
The LMNA gene encodes lamins A and C, two intermediate filament-type proteins that are important determinants of interphase nuclear architecture. Mutations in LMNA lead to a wide spectrum of human diseases including autosomal dominant Emery-Dreifuss muscular dystrophy (AD-EDMD), which affects skeletal and cardiac muscle. The cellular mechanisms by which mutations in LMNA cause disease have been elusive. Here, we demonstrate that defects in neuromuscular junctions (NMJs) are part of the disease mechanism in AD-EDMD. Two AD-EDMD mouse models show innervation defects including misexpression of electrical activity-dependent genes and altered epigenetic chromatin modifications. Synaptic nuclei are not properly recruited to the NMJ because of mislocalization of nuclear envelope components. AD-EDMD patients with LMNA mutations show the same cellular defects as the AD-EDMD mouse models. These results suggest that lamin A/C-mediated NMJ defects contribute to the AD-EDMD disease phenotype and provide insights into the cellular and molecular mechanisms for the muscle-specific phenotype of AD-EDMD.
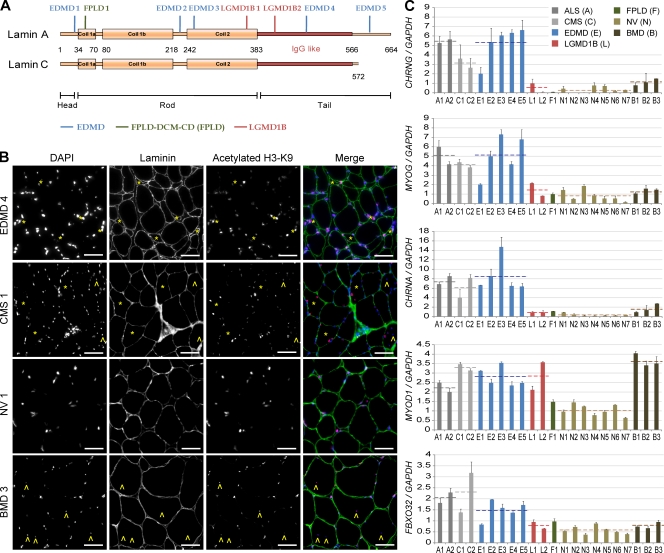
Figures


Similar articles
-
Uncoordinated transcription and compromised muscle function in the lmna-null mouse model of Emery- Emery-Dreyfuss muscular dystrophy.PLoS One. 2011 Feb 22;6(2):e16651. doi: 10.1371/journal.pone.0016651. PLoS One. 2011. PMID: 21364987 Free PMC article.
-
Expression and localization of nuclear proteins in autosomal-dominant Emery-Dreifuss muscular dystrophy with LMNA R377H mutation.BMC Cell Biol. 2004 Mar 30;5:12. doi: 10.1186/1471-2121-5-12. BMC Cell Biol. 2004. PMID: 15053843 Free PMC article.
-
Lamin A/C Assembly Defects in LMNA-Congenital Muscular Dystrophy Is Responsible for the Increased Severity of the Disease Compared with Emery-Dreifuss Muscular Dystrophy.Cells. 2020 Mar 31;9(4):844. doi: 10.3390/cells9040844. Cells. 2020. PMID: 32244403 Free PMC article.
-
Emery-Dreifuss muscular dystrophy: focal point nuclear envelope.Curr Opin Neurol. 2019 Oct;32(5):728-734. doi: 10.1097/WCO.0000000000000741. Curr Opin Neurol. 2019. PMID: 31460960 Free PMC article. Review.
-
Emery-Dreifuss muscular dystrophy.Curr Neurol Neurosci Rep. 2007 Jan;7(1):78-83. doi: 10.1007/s11910-007-0025-3. Curr Neurol Neurosci Rep. 2007. PMID: 17217858 Review.
Cited by
-
Uncoordinated transcription and compromised muscle function in the lmna-null mouse model of Emery- Emery-Dreyfuss muscular dystrophy.PLoS One. 2011 Feb 22;6(2):e16651. doi: 10.1371/journal.pone.0016651. PLoS One. 2011. PMID: 21364987 Free PMC article.
-
Axonal regeneration and neuronal function are preserved in motor neurons lacking ß-actin in vivo.PLoS One. 2011 Mar 22;6(3):e17768. doi: 10.1371/journal.pone.0017768. PLoS One. 2011. PMID: 21445349 Free PMC article.
-
Moving and positioning the nucleus in skeletal muscle - one step at a time.Nucleus. 2015;6(5):373-81. doi: 10.1080/19491034.2015.1090073. Nucleus. 2015. PMID: 26338260 Free PMC article. Review.
-
Lamin A/C Cardiomyopathies: Current Understanding and Novel Treatment Strategies.Curr Treat Options Cardiovasc Med. 2017 Mar;19(3):21. doi: 10.1007/s11936-017-0520-z. Curr Treat Options Cardiovasc Med. 2017. PMID: 28299614 Review.
-
The diverse functional LINCs of the nuclear envelope to the cytoskeleton and chromatin.Chromosoma. 2013 Oct;122(5):415-29. doi: 10.1007/s00412-013-0417-x. Epub 2013 Jun 5. Chromosoma. 2013. PMID: 23736899 Free PMC article. Review.
References
-
- Apel, E.D., R.M. Lewis, R.M. Grady, and J.R. Sanes. 2000. Syne-1, a dystrophin- and Klarsicht-related protein associated with synaptic nuclei at the neuromuscular junction. J. Biol. Chem. 275:31986–31995. - PubMed
-
- Arimura, T., A. Helbling-Leclerc, C. Massart, S. Varnous, F. Niel, E. Lacene, Y. Fromes, M. Toussaint, A.M. Mura, D.I. Keller, et al. 2005. Mouse model carrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum. Mol. Genet. 14:155–169. - PubMed
-
- Bakay, M., Z. Wang, G. Melcon, L. Schiltz, J. Xuan, P. Zhao, V. Sartorelli, J. Seo, E. Pegoraro, C. Angelini, et al. 2006. Nuclear envelope dystrophies show a transcriptional fingerprint suggesting disruption of Rb-MyoD pathways in muscle regeneration. Brain. 129:996–1013. - PubMed
-
- Bhagavati, S., A. Ghatpande, S.A. Shafiq, and B. Leung. 1996. In situ hybridization analysis for expression of myogenic regulatory factors in regenerating muscle of mdx mouse. J. Neuropathol. Exp. Neurol. 55:509–514. - PubMed
-
- Broers, J.L., E.A. Peeters, H.J. Kuijpers, J. Endert, C.V. Bouten, C.W. Oomens, F.P. Baaijens, and F.C. Ramaekers. 2004. Decreased mechanical stiffness in LMNA−/− cells is caused by defective nucleo-cytoskeletal integrity: implications for the development of laminopathies. Hum. Mol. Genet. 13:2567–2580. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
