

WGCNA: an R package for weighted correlation network analysis
- PMID: 19114008
- PMCID: PMC2631488
- DOI: 10.1186/1471-2105-9-559
WGCNA: an R package for weighted correlation network analysis
Abstract
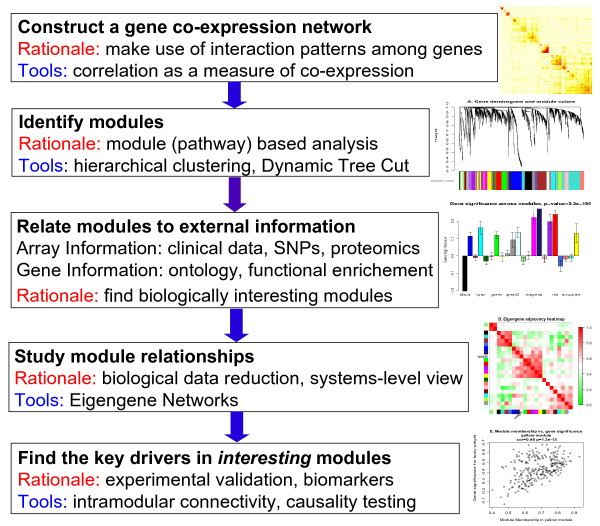
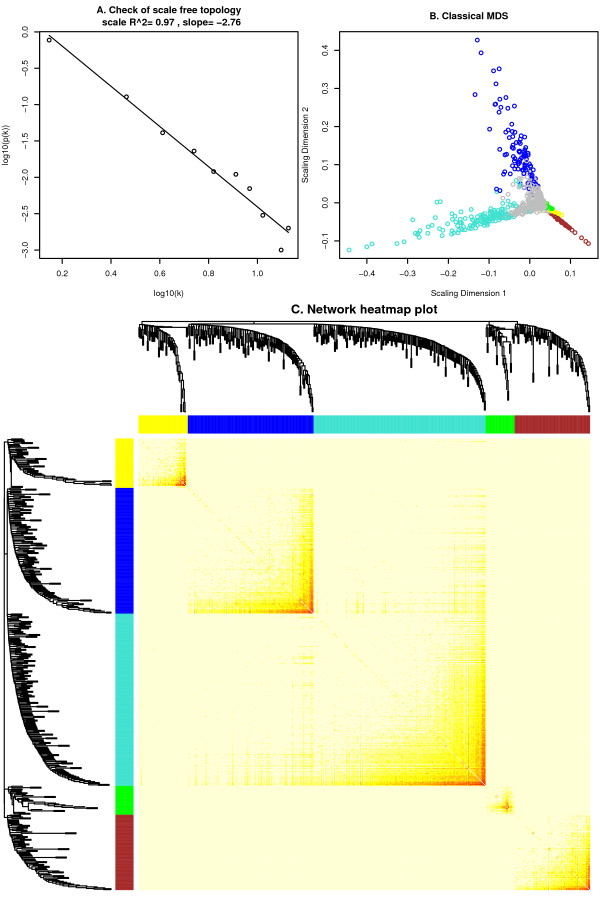
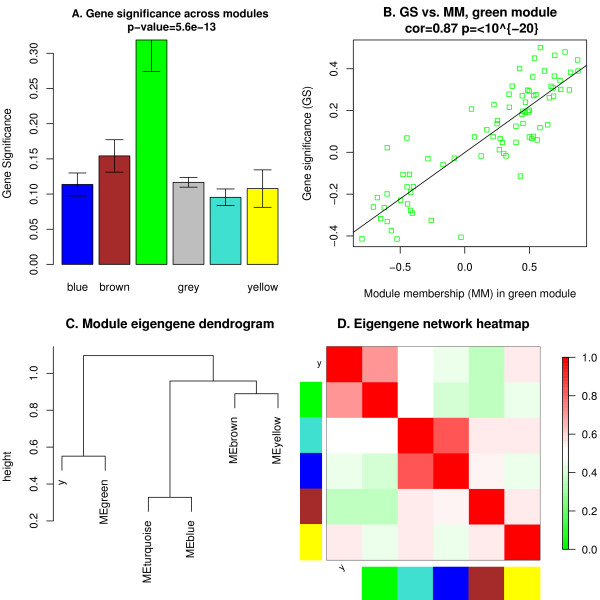
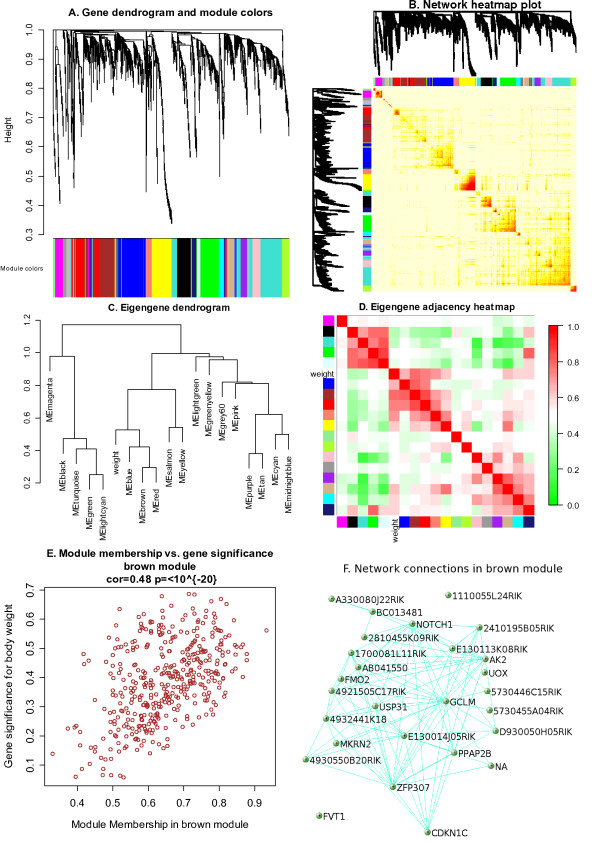
Background: Correlation networks are increasingly being used in bioinformatics applications. For example, weighted gene co-expression network analysis is a systems biology method for describing the correlation patterns among genes across microarray samples. Weighted correlation network analysis (WGCNA) can be used for finding clusters (modules) of highly correlated genes, for summarizing such clusters using the module eigengene or an intramodular hub gene, for relating modules to one another and to external sample traits (using eigengene network methodology), and for calculating module membership measures. Correlation networks facilitate network based gene screening methods that can be used to identify candidate biomarkers or therapeutic targets. These methods have been successfully applied in various biological contexts, e.g. cancer, mouse genetics, yeast genetics, and analysis of brain imaging data. While parts of the correlation network methodology have been described in separate publications, there is a need to provide a user-friendly, comprehensive, and consistent software implementation and an accompanying tutorial.
Results: The WGCNA R software package is a comprehensive collection of R functions for performing various aspects of weighted correlation network analysis. The package includes functions for network construction, module detection, gene selection, calculations of topological properties, data simulation, visualization, and interfacing with external software. Along with the R package we also present R software tutorials. While the methods development was motivated by gene expression data, the underlying data mining approach can be applied to a variety of different settings.
Conclusion: The WGCNA package provides R functions for weighted correlation network analysis, e.g. co-expression network analysis of gene expression data. The R package along with its source code and additional material are freely available at http://www.genetics.ucla.edu/labs/horvath/CoexpressionNetwork/Rpackages/WGCNA.
Figures
Similar articles
-
Eigengene networks for studying the relationships between co-expression modules.BMC Syst Biol. 2007 Nov 21;1:54. doi: 10.1186/1752-0509-1-54. BMC Syst Biol. 2007. PMID: 18031580 Free PMC article.
-
SPRINT: a new parallel framework for R.BMC Bioinformatics. 2008 Dec 29;9:558. doi: 10.1186/1471-2105-9-558. BMC Bioinformatics. 2008. PMID: 19114001 Free PMC article.
-
Weighted gene co-expression network analysis identifies specific modules and hub genes related to coronary artery disease.BMC Cardiovasc Disord. 2016 Mar 5;16:54. doi: 10.1186/s12872-016-0217-3. BMC Cardiovasc Disord. 2016. PMID: 26944061 Free PMC article.
-
Exploiting systems biology to investigate the gene modules and drugs in ovarian cancer: A hypothesis based on the weighted gene co-expression network analysis.Biomed Pharmacother. 2022 Feb;146:112537. doi: 10.1016/j.biopha.2021.112537. Epub 2021 Dec 16. Biomed Pharmacother. 2022. PMID: 34922114 Review.
-
Matrix factorisation methods applied in microarray data analysis.Int J Data Min Bioinform. 2010;4(1):72-90. doi: 10.1504/ijdmb.2010.030968. Int J Data Min Bioinform. 2010. PMID: 20376923 Free PMC article. Review.
Cited by
-
Comparative Transcriptomic Analysis Reveals Domestication and Improvement Patterns of Broomcorn Millet (Panicum miliaceum L.).Int J Mol Sci. 2024 Oct 13;25(20):11012. doi: 10.3390/ijms252011012. Int J Mol Sci. 2024. PMID: 39456795 Free PMC article.
-
Quantitative Trait Locus Mapping Combined with RNA Sequencing Identified Candidate Genes for Resistance to Powdery Mildew in Bitter Gourd (Momordica charantia L.).Int J Mol Sci. 2024 Oct 15;25(20):11080. doi: 10.3390/ijms252011080. Int J Mol Sci. 2024. PMID: 39456862 Free PMC article.
-
Multiple Machine Learning Identifies Key Gene PHLDA1 Suppressing NAFLD Progression.Inflammation. 2024 Nov 4. doi: 10.1007/s10753-024-02164-6. Online ahead of print. Inflammation. 2024. PMID: 39496918
-
Identification of a seven-gene prognostic model for renal cell carcinoma associated with CD8+T lymphocyte cell.Medicine (Baltimore). 2024 Oct 4;103(40):e39938. doi: 10.1097/MD.0000000000039938. Medicine (Baltimore). 2024. PMID: 39465721 Free PMC article.
-
UV-induced reactive oxygen species and transcriptional control of 3-deoxyanthocyanidin biosynthesis in black sorghum pericarp.Front Plant Sci. 2024 Oct 7;15:1451215. doi: 10.3389/fpls.2024.1451215. eCollection 2024. Front Plant Sci. 2024. PMID: 39435026 Free PMC article.
References
-
- Fisher RA. On the 'probable error' of a coefficient of correlation deduced from a small sample. Metron. 1915;1:1–32.
-
- Stuart JM, Segal E, Koller D, Kim SK. A Gene-Coexpression Network for Global Discovery of Conserved Genetic Modules. Science. 2003;302:249–255. - PubMed
-
- Zhang B, Horvath S. A General Framework for Weighted Gene Co-expression Network Analysis. Stat Appl Genet Mol Biol. 2005;4:Article 17. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
