

De novo protein structure generation from incomplete chemical shift assignments
- PMID: 19034676
- PMCID: PMC2683404
- DOI: 10.1007/s10858-008-9288-5
De novo protein structure generation from incomplete chemical shift assignments
Abstract
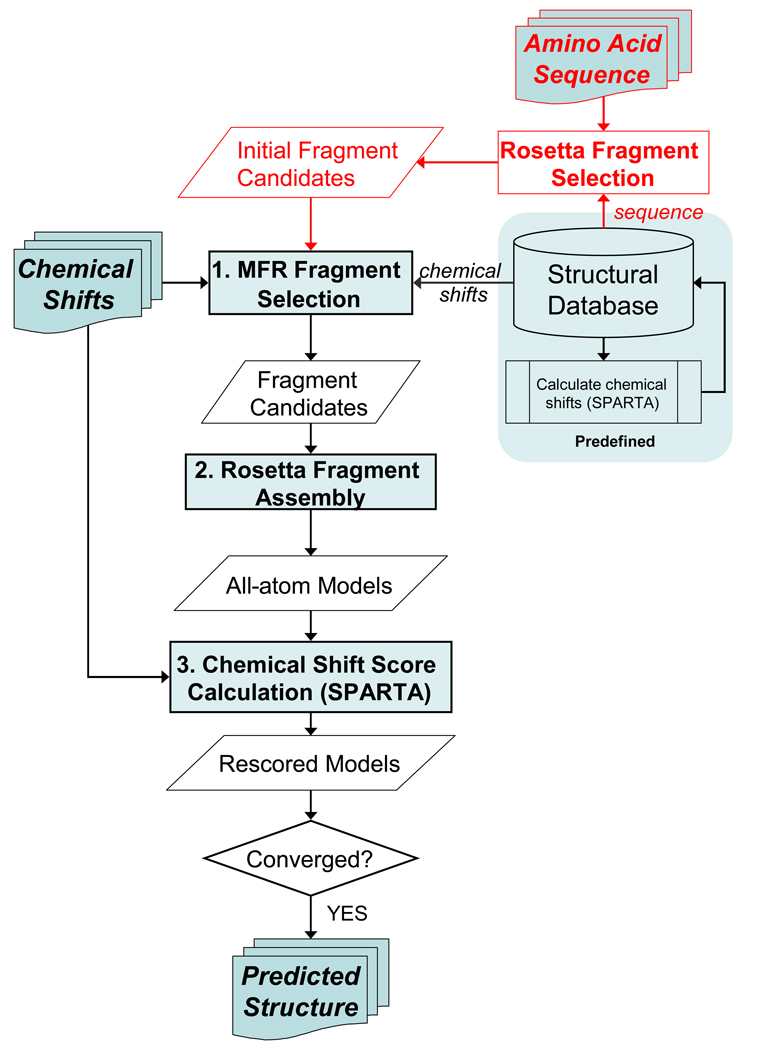
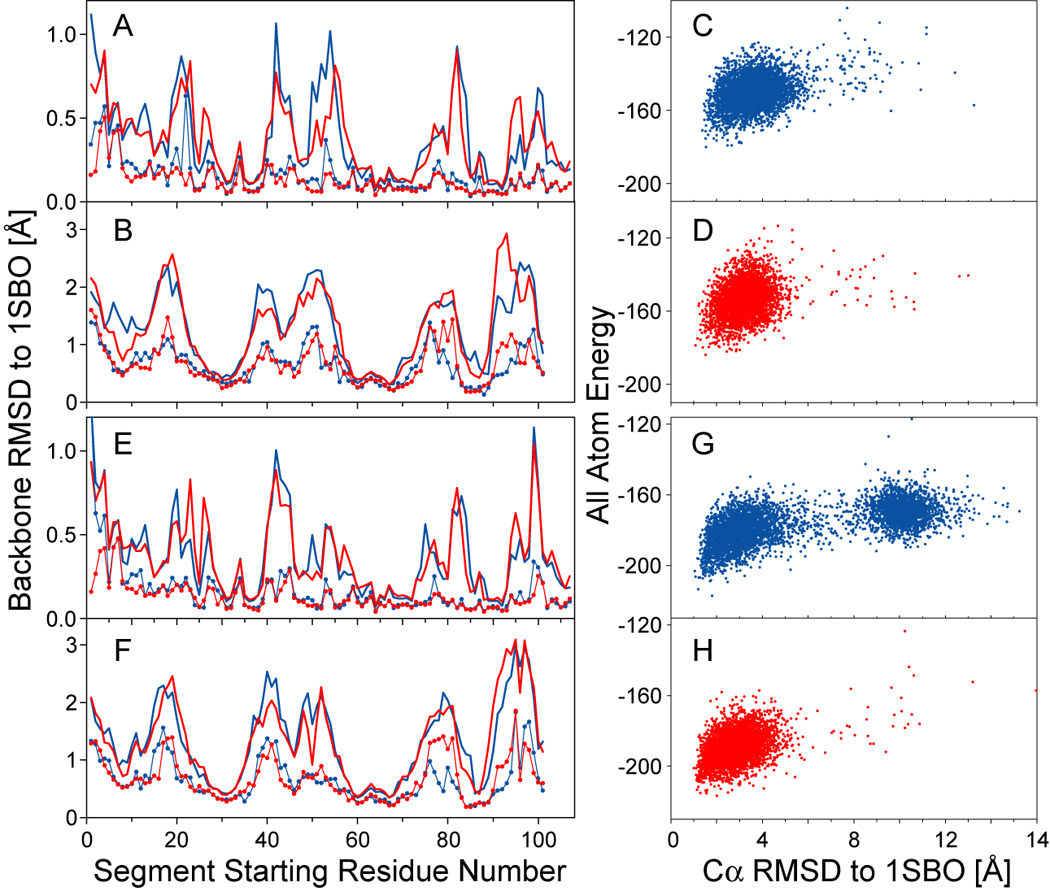
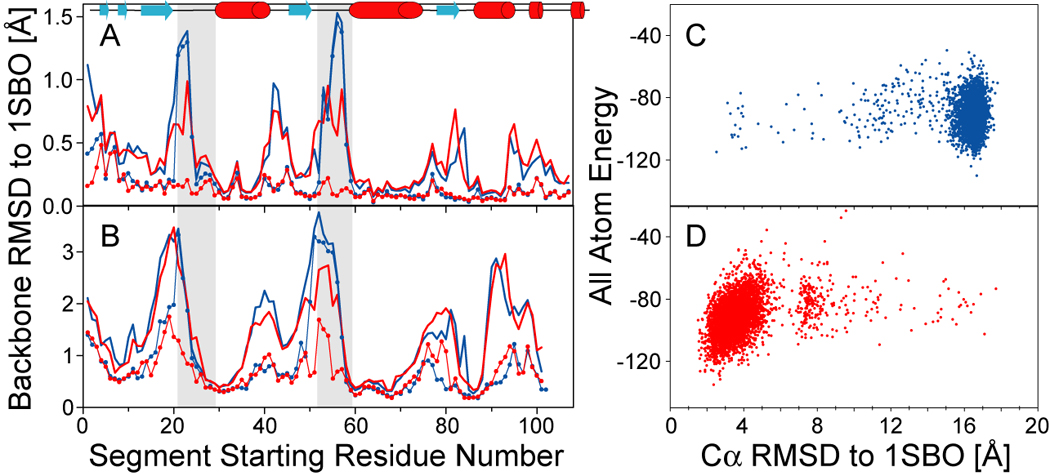
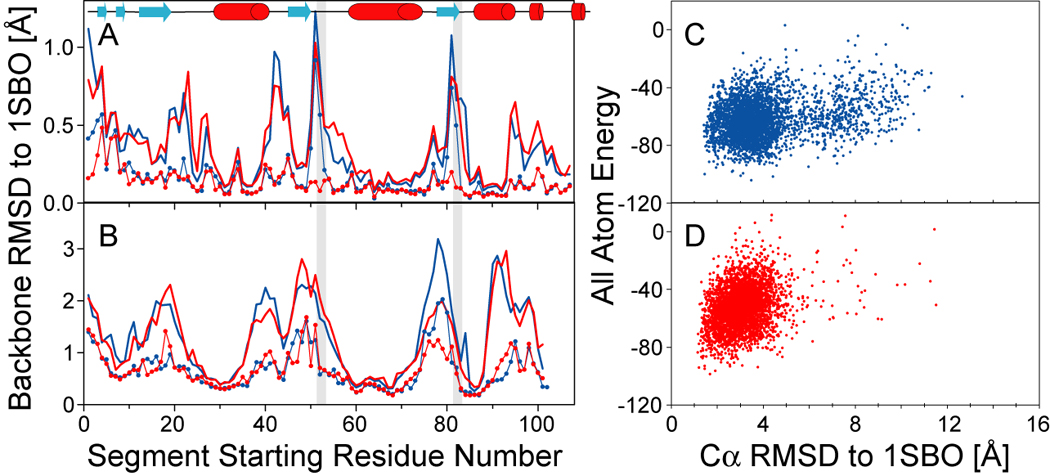
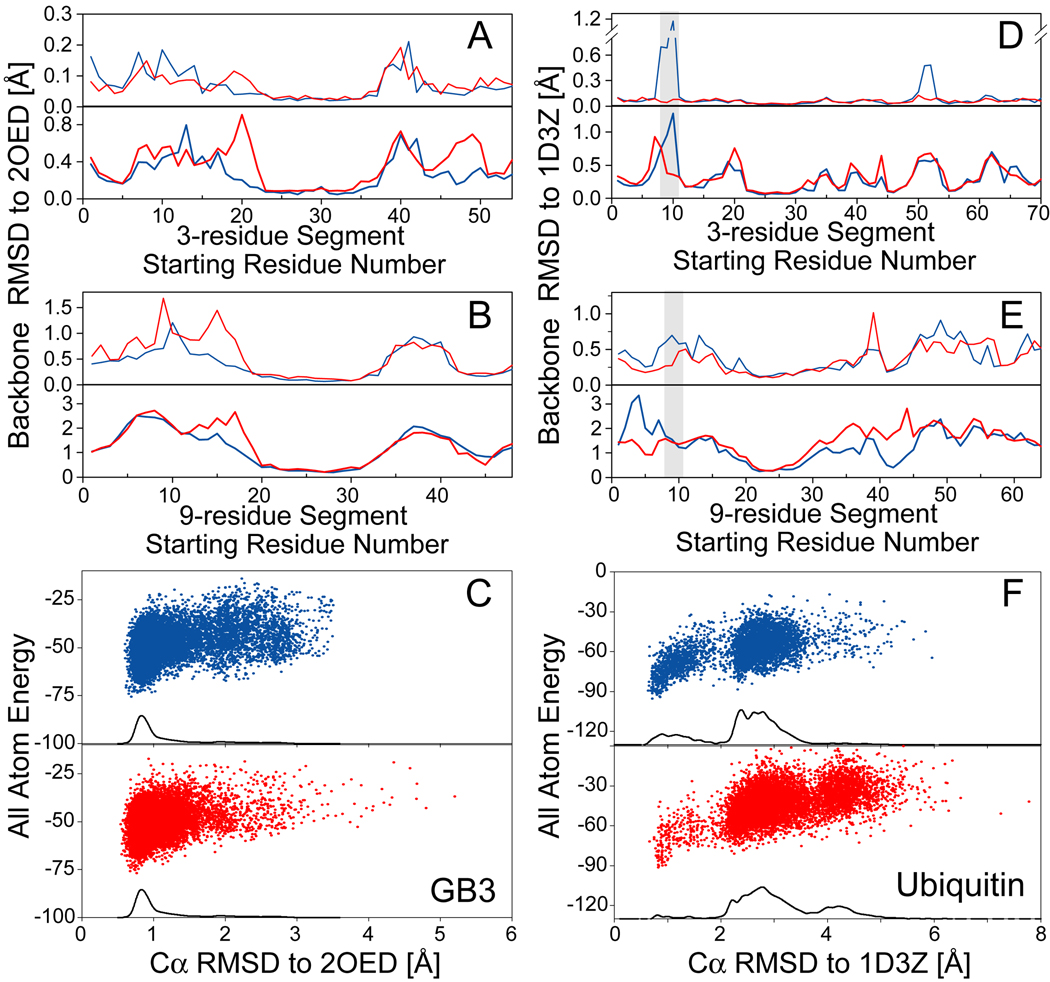
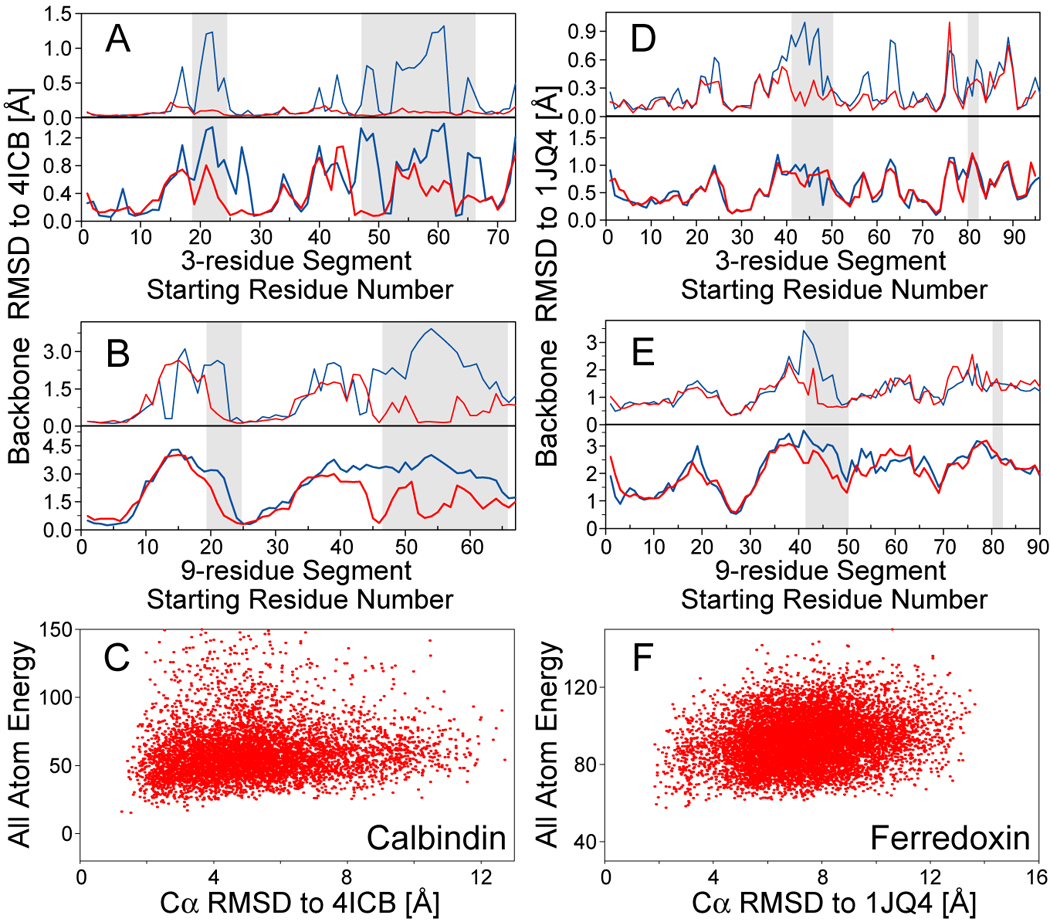
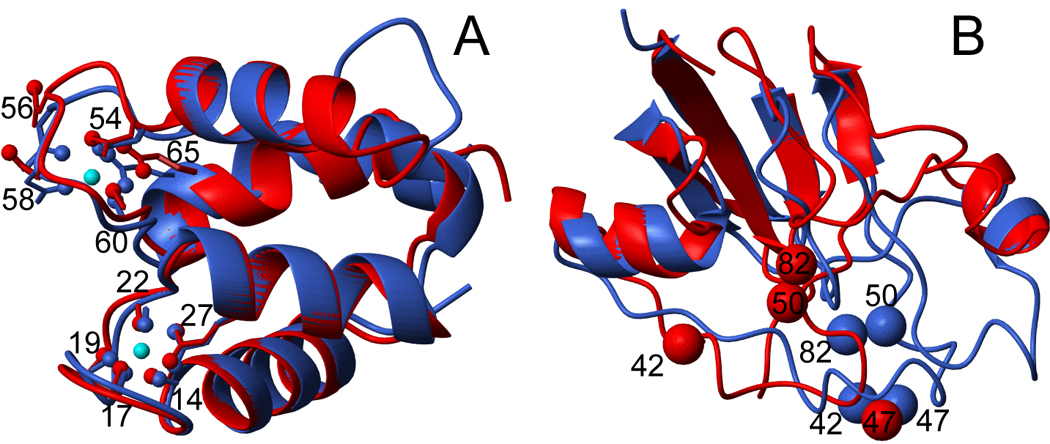
NMR chemical shifts provide important local structural information for proteins. Consistent structure generation from NMR chemical shift data has recently become feasible for proteins with sizes of up to 130 residues, and such structures are of a quality comparable to those obtained with the standard NMR protocol. This study investigates the influence of the completeness of chemical shift assignments on structures generated from chemical shifts. The Chemical-Shift-Rosetta (CS-Rosetta) protocol was used for de novo protein structure generation with various degrees of completeness of the chemical shift assignment, simulated by omission of entries in the experimental chemical shift data previously used for the initial demonstration of the CS-Rosetta approach. In addition, a new CS-Rosetta protocol is described that improves robustness of the method for proteins with missing or erroneous NMR chemical shift input data. This strategy, which uses traditional Rosetta for pre-filtering of the fragment selection process, is demonstrated for two paramagnetic proteins and also for two proteins with solid-state NMR chemical shift assignments.
Figures
Similar articles
-
Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA.J Mol Biol. 2002 May 24;319(1):209-27. doi: 10.1016/s0022-2836(02)00241-3. J Mol Biol. 2002. PMID: 12051947
-
Influence of the completeness of chemical shift assignments on NMR structures obtained with automated NOE assignment.J Struct Funct Genomics. 2003;4(2-3):179-89. doi: 10.1023/a:1026122726574. J Struct Funct Genomics. 2003. PMID: 14649302
-
Improved chemical shift based fragment selection for CS-Rosetta using Rosetta3 fragment picker.J Biomol NMR. 2013 Oct;57(2):117-27. doi: 10.1007/s10858-013-9772-4. Epub 2013 Aug 22. J Biomol NMR. 2013. PMID: 23975356
-
Recent Advances in NMR Protein Structure Prediction with ROSETTA.Int J Mol Sci. 2023 Apr 25;24(9):7835. doi: 10.3390/ijms24097835. Int J Mol Sci. 2023. PMID: 37175539 Free PMC article. Review.
-
Automation of NMR structure determination of proteins.Curr Opin Struct Biol. 2004 Oct;14(5):547-53. doi: 10.1016/j.sbi.2004.09.003. Curr Opin Struct Biol. 2004. PMID: 15465314 Review.
Cited by
-
High resolution structural characterization of Aβ42 amyloid fibrils by magic angle spinning NMR.J Am Chem Soc. 2015 Jun 17;137(23):7509-18. doi: 10.1021/jacs.5b03997. Epub 2015 Jun 4. J Am Chem Soc. 2015. PMID: 26001057 Free PMC article.
-
Protein fold determined by paramagnetic magic-angle spinning solid-state NMR spectroscopy.Nat Chem. 2012 Mar 18;4(5):410-7. doi: 10.1038/nchem.1299. Nat Chem. 2012. PMID: 22522262 Free PMC article.
-
PEA-15 C-Terminal Tail Allosterically Modulates Death-Effector Domain Conformation and Facilitates Protein-Protein Interactions.Int J Mol Sci. 2019 Jul 7;20(13):3335. doi: 10.3390/ijms20133335. Int J Mol Sci. 2019. PMID: 31284641 Free PMC article.
-
Density functional calculations of backbone 15N shielding tensors in beta-sheet and turn residues of protein G.J Biomol NMR. 2011 May;50(1):19-33. doi: 10.1007/s10858-011-9474-8. Epub 2011 Feb 9. J Biomol NMR. 2011. PMID: 21305337 Free PMC article.
-
An automated framework for NMR resonance assignment through simultaneous slice picking and spin system forming.J Biomol NMR. 2014 Jun;59(2):75-86. doi: 10.1007/s10858-014-9828-0. Epub 2014 Apr 19. J Biomol NMR. 2014. PMID: 24748536
References
-
- Agarwal V, Diehl A, Skrynnikov N, Reif B. High resolution H-1 detected H-1,C-13 correlation spectra in MAS solid-state NMR using deuterated proteins with selective H-1,H-2 isotopic labeling of methyl groups. J. Am. Chem. Soc. 2006;128:12620–12621. - PubMed
-
- Ando I, Kameda T, Asakawa N, Kuroki S, Kurosu H. Structure of peptides and polypeptides in the solid state as elucidated by NMR chemical shift. J. Mol. Struct. 1998;441:213–230.
-
- Andreini C, Bertini I, Rosato A. A hint to search for metalloproteins in gene banks. Bioinformatics. 2004;20:1373–1380. - PubMed
-
- Asakura T, Demura M, Date T, Miyashita N, Ogawa K, Williamson MP. NMR study of silk I structure of Bombyx mori silk fibroin with N-15- and C-13-NMR chemical shift contour plots. Biopolymers. 1997;41:193–203.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
