

Disruption of nesprin-1 produces an Emery Dreifuss muscular dystrophy-like phenotype in mice
- PMID: 19008300
- PMCID: PMC2722216
- DOI: 10.1093/hmg/ddn386
Disruption of nesprin-1 produces an Emery Dreifuss muscular dystrophy-like phenotype in mice
Abstract
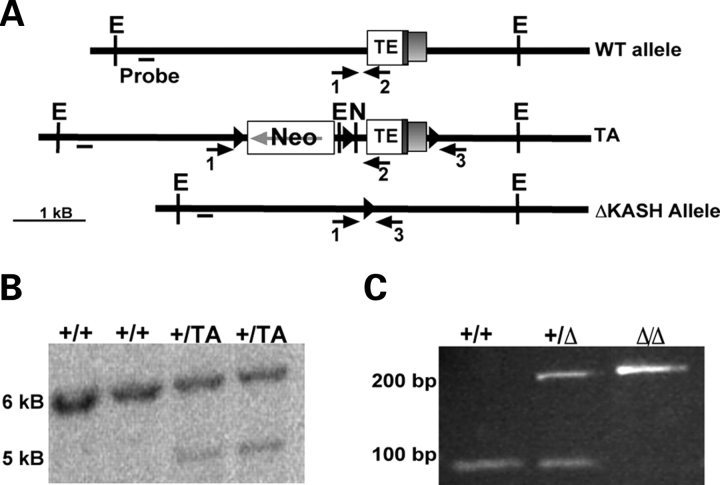
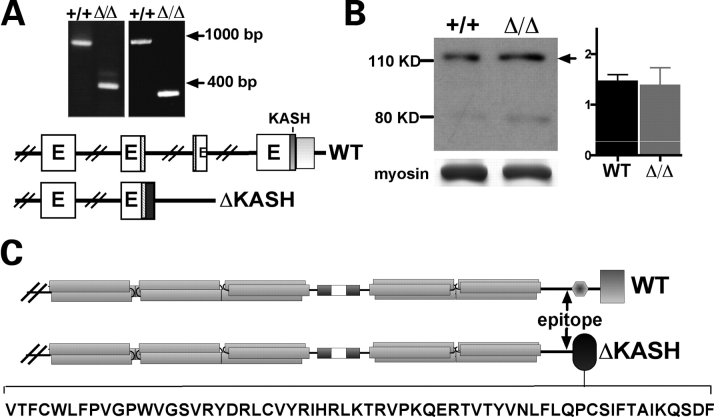
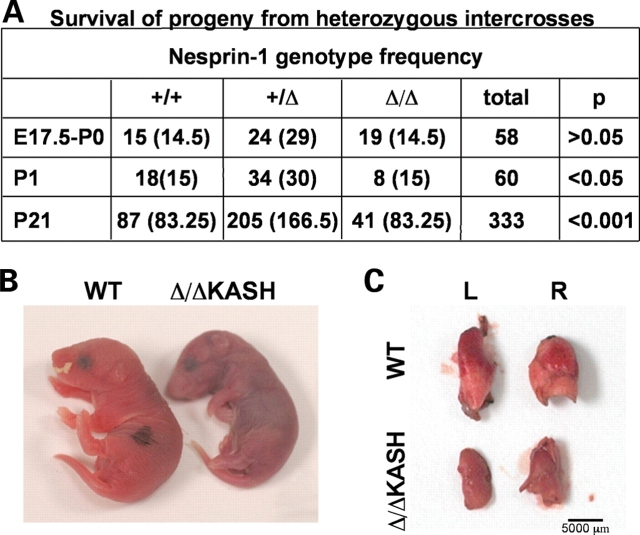
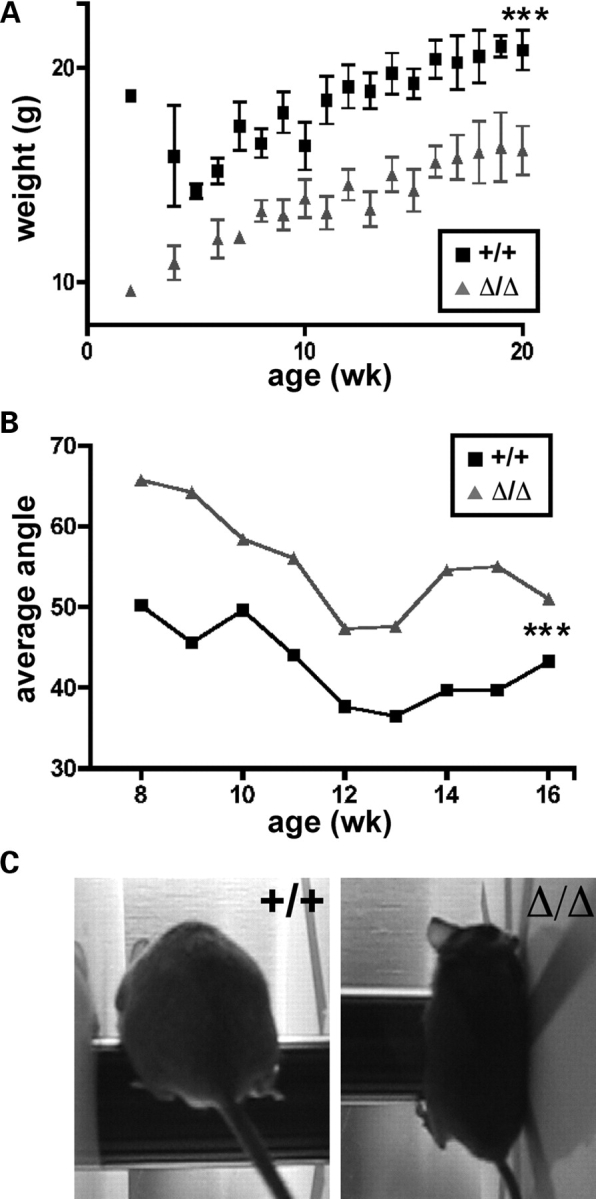
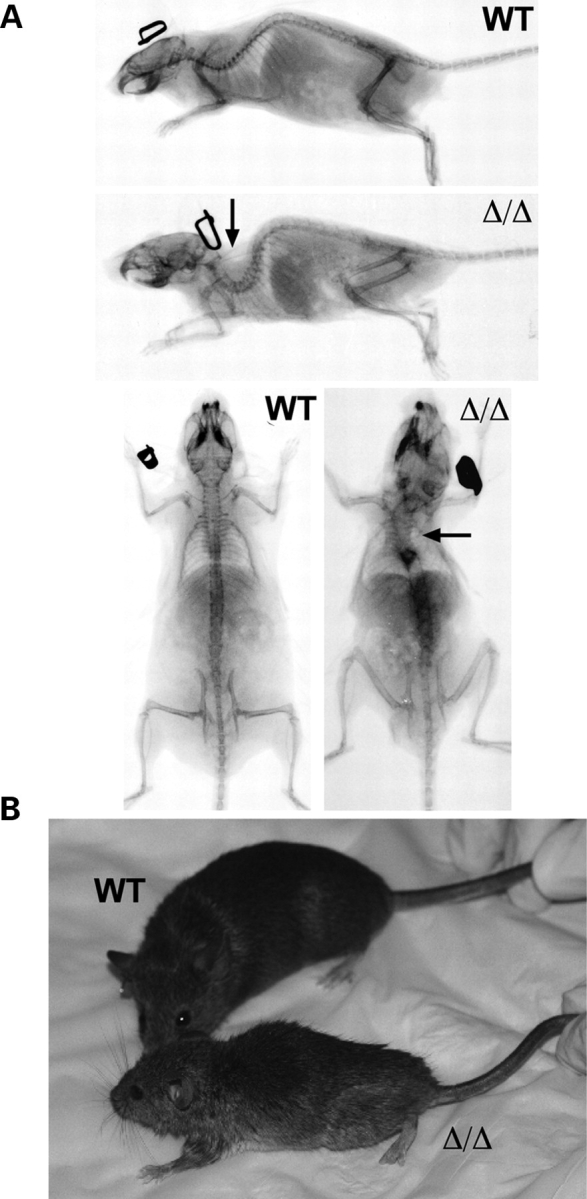
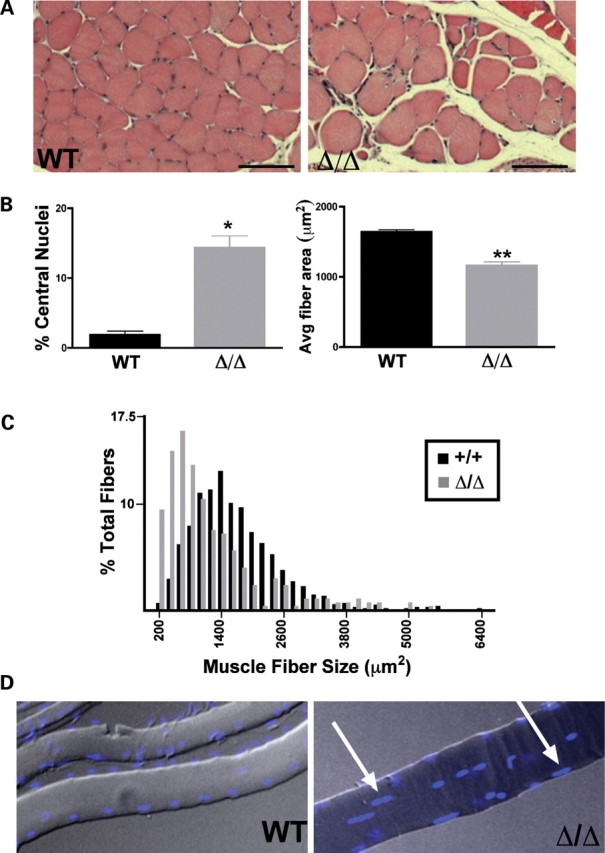
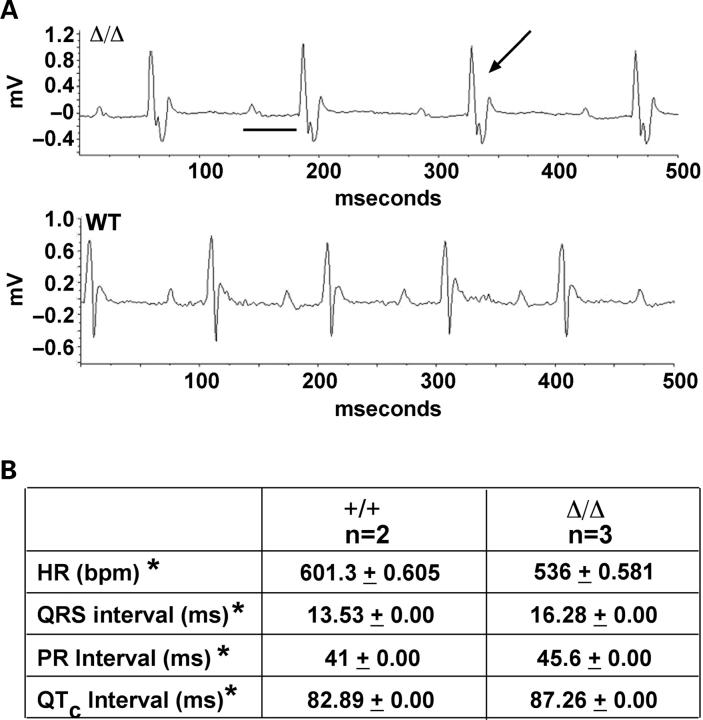
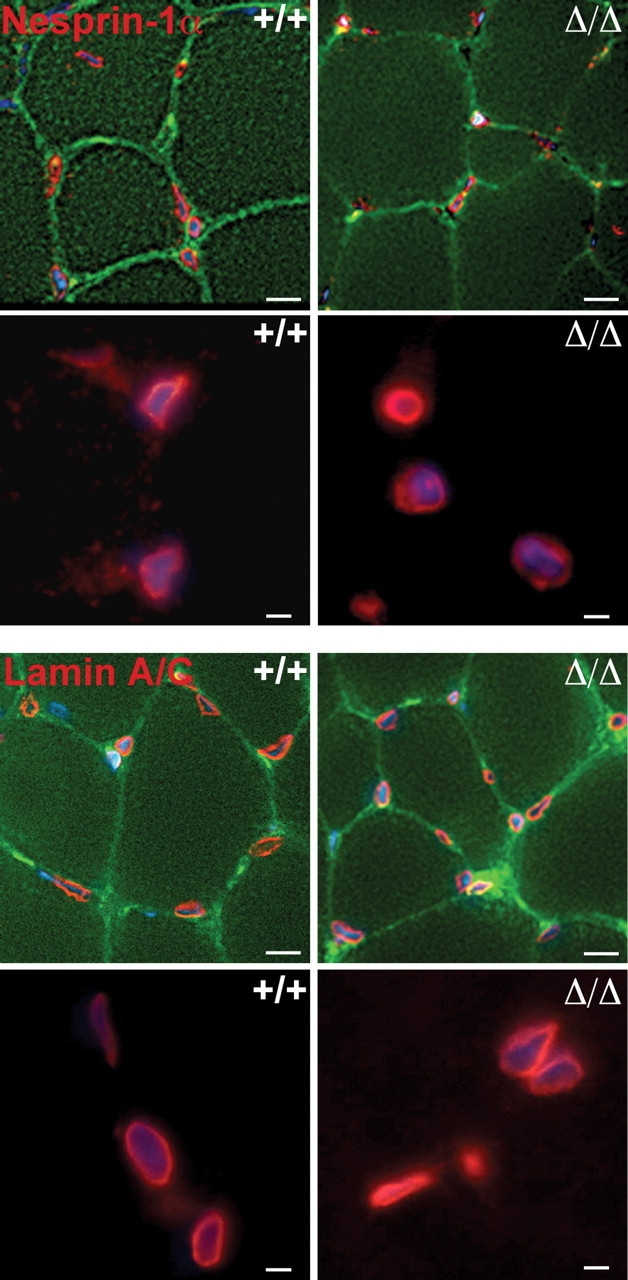
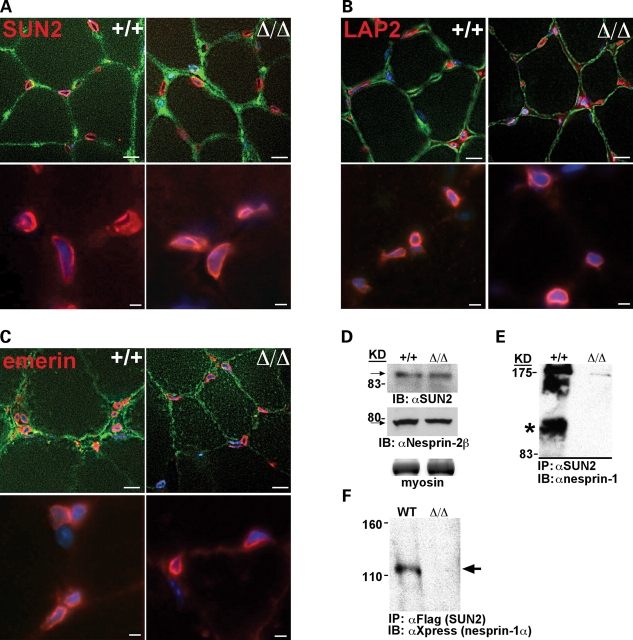
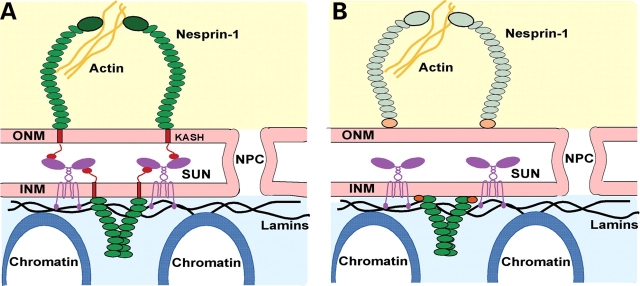
Mutations in the gene encoding the inner nuclear membrane proteins lamins A and C produce cardiac and skeletal muscle dysfunction referred to as Emery Dreifuss muscular dystrophy. Lamins A and C participate in the LINC complex that, along with the nesprin and SUN proteins, LInk the Nucleoskeleton with the Cytoskeleton. Nesprins 1 and 2 are giant spectrin-repeat containing proteins that have large and small forms. The nesprins contain a transmembrane anchor that tethers to the nuclear membrane followed by a short domain that resides within the lumen between the inner and outer nuclear membrane. Nesprin's luminal domain binds directly to SUN proteins. We generated mice where the C-terminus of nesprin-1 was deleted. This strategy produced a protein lacking the transmembrane and luminal domains that together are referred to as the KASH domain. Mice homozygous for this mutation exhibit lethality with approximately half dying at or near birth from respiratory failure. Surviving mice display hindlimb weakness and an abnormal gait. With increasing age, kyphoscoliosis, muscle pathology and cardiac conduction defects develop. The protein components of the LINC complex, including mutant nesprin-1alpha, lamin A/C and SUN2, are localized at the nuclear membrane in this model. However, the LINC components do not normally associate since coimmunoprecipitation experiments with SUN2 and nesprin reveal that mutant nesprin-1 protein no longer interacts with SUN2. These findings demonstrate the role of the LINC complex, and nesprin-1, in neuromuscular and cardiac disease.
Figures
Similar articles
-
Nesprin-1 and -2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity.Hum Mol Genet. 2007 Dec 1;16(23):2816-33. doi: 10.1093/hmg/ddm238. Epub 2007 Aug 29. Hum Mol Genet. 2007. PMID: 17761684
-
Novel nesprin-1 mutations associated with dilated cardiomyopathy cause nuclear envelope disruption and defects in myogenesis.Hum Mol Genet. 2017 Jun 15;26(12):2258-2276. doi: 10.1093/hmg/ddx116. Hum Mol Genet. 2017. PMID: 28398466 Free PMC article.
-
Nesprin 1 is critical for nuclear positioning and anchorage.Hum Mol Genet. 2010 Jan 15;19(2):329-41. doi: 10.1093/hmg/ddp499. Epub 2009 Oct 28. Hum Mol Genet. 2010. PMID: 19864491 Free PMC article.
-
Nesprin proteins: bridging nuclear envelope dynamics to muscular dysfunction.Cell Commun Signal. 2024 Apr 2;22(1):208. doi: 10.1186/s12964-024-01593-y. Cell Commun Signal. 2024. PMID: 38566066 Free PMC article. Review.
-
LINC complexes in health and disease.Nucleus. 2010 Jan-Feb;1(1):40-52. doi: 10.4161/nucl.1.1.10530. Nucleus. 2010. PMID: 21327104 Free PMC article. Review.
Cited by
-
Molecular mechanisms of centrosome and cytoskeleton anchorage at the nuclear envelope.Cell Mol Life Sci. 2011 May;68(9):1593-610. doi: 10.1007/s00018-010-0535-z. Epub 2010 Oct 5. Cell Mol Life Sci. 2011. PMID: 20922455 Free PMC article.
-
SYNE1-related autosomal recessive cerebellar ataxia, congenital cerebellar hypoplasia, and cognitive impairment.Clin Pract. 2018 Aug 27;8(3):1071. doi: 10.4081/cp.2018.1071. eCollection 2018 Jul 10. Clin Pract. 2018. PMID: 30275942 Free PMC article.
-
Nesprin-1 LINC complexes recruit microtubule cytoskeleton proteins and drive pathology in Lmna-mutant striated muscle.Hum Mol Genet. 2023 Jan 6;32(2):177-191. doi: 10.1093/hmg/ddac179. Hum Mol Genet. 2023. PMID: 35925868 Free PMC article.
-
Drosophila Nesprin-1 Isoforms Differentially Contribute to Muscle Function.Cells. 2021 Nov 6;10(11):3061. doi: 10.3390/cells10113061. Cells. 2021. PMID: 34831284 Free PMC article.
-
The Drosophila Nesprin-1 homolog MSP300 is required for muscle autophagy and proteostasis.J Cell Sci. 2024 Jun 1;137(11):jcs262096. doi: 10.1242/jcs.262096. Epub 2024 Jun 10. J Cell Sci. 2024. PMID: 38757366 Free PMC article.
References
-
- Favreau C., Dubosclard E., Ostlund C., Vigouroux C., Capeau J., Wehnert M., Higuet D., Worman H.J., Courvalin J.C., Buendia B. Expression of lamin A mutated in the carboxyl-terminal tail generates an aberrant nuclear phenotype similar to that observed in cells from patients with Dunnigan-type partial lipodystrophy and Emery-Dreifuss muscular dystrophy. Exp. Cell Res. 2003;282:14–23. - PubMed
-
- Bonne G., Mercuri E., Muchir A., Urtizberea A., Becane H.M., Recan D., Merlini L., Wehnert M., Boor R., Reuner U., et al. Clinical and molecular genetic spectrum of autosomal dominant Emery-Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann. Neurol. 2000;48:170–180. - PubMed
-
- Wang Y., Herron A.J., Worman H.J. Pathology and nuclear abnormalities in hearts of transgenic mice expressing M371K lamin A encoded by an LMNA mutation causing Emery-Dreifuss muscular dystrophy. Hum. Mol. Genet. 2006;15:2479–2489. - PubMed
-
- Arimura T., Helbling-Leclerc A., Massart C., Varnous S., Niel F., Lacene E., Fromes Y., Toussaint M., Mura A.M., Keller D.I., et al. Mouse model carrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum. Mol. Genet. 2005;14:155–169. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
