

Lung alveolar integrity is compromised by telomere shortening in telomerase-null mice
- PMID: 18952756
- PMCID: PMC2636955
- DOI: 10.1152/ajplung.90411.2008
Lung alveolar integrity is compromised by telomere shortening in telomerase-null mice
Abstract
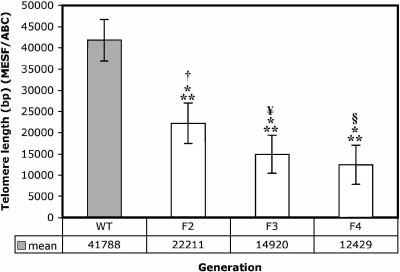
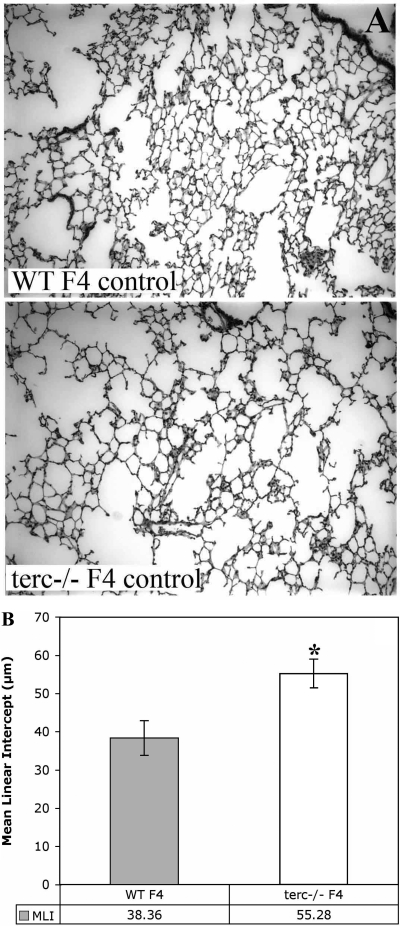
Shortened telomeres are a normal consequence of cell division. However, telomere shortening past a critical point results in cellular senescence and death. To determine the effect of telomere shortening on lung, four generations of B6.Cg-Terc(tm1Rdp) mice, null for the terc component of telomerase, the holoenzyme that maintains telomeres, were bred and analyzed. Generational inbreeding of terc-/- mice caused sequential shortening of telomeres. Lung histology from the generation with the shortest telomeres (terc-/- F4) showed alveolar wall thinning and increased alveolar size. Morphometric analysis confirmed a significant increase in mean linear intercept (MLI). terc-/- F4 lung showed normal elastin deposition but had significantly decreased collagen content. Both airway and alveolar epithelial type 1 cells (AEC1) appeared normal by immunohistochemistry, and the percentage of alveolar epithelial type 2 cells (AEC2) per total cell number was similar to wild type. However, because of a decrease in distal lung cellularity, the absolute number of AEC2 in terc-/- F4 lung was significantly reduced. In contrast to wild type, terc-/- F4 distal lung epithelium from normoxia-maintained mice exhibited DNA damage by terminal deoxynucleotidyltransferase (TdT)-mediated dUTP nick end labeling (TUNEL) and 8-oxoguanine immunohistochemistry. Western blotting of freshly isolated AEC2 lysates for stress signaling kinases confirmed that the stress-activated protein kinase (SAPK)/c-Jun NH(2)-terminal kinase (JNK) stress response pathway is stimulated in telomerase-null AEC2 even under normoxic conditions. Expression of downstream apoptotic/stress markers, including caspase-3, caspase-6, Bax, and HSP-25, was also observed in telomerase-null, but not wild-type, AEC2. TUNEL analysis of freshly isolated normoxic AEC2 showed that DNA strand breaks, essentially absent in wild-type cells, increased with each successive terc-/- generation and correlated strongly with telomere length (R(2) = 0.9631). Thus lung alveolar integrity, particularly in the distal epithelial compartment, depends on proper telomere maintenance.
Figures







Similar articles
-
Telomerase deficiency does not alter bleomycin-induced fibrosis in mice.Exp Lung Res. 2012 Apr;38(3):124-34. doi: 10.3109/01902148.2012.658148. Exp Lung Res. 2012. PMID: 22394286 Free PMC article.
-
Pulmonary Alveolar Stem Cell Senescence, Apoptosis, and Differentiation by p53-Dependent and -Independent Mechanisms in Telomerase-Deficient Mice.Cells. 2021 Oct 26;10(11):2892. doi: 10.3390/cells10112892. Cells. 2021. PMID: 34831112 Free PMC article.
-
Partial pneumonectomy of telomerase null mice carrying shortened telomeres initiates cell growth arrest resulting in a limited compensatory growth response.Am J Physiol Lung Cell Mol Physiol. 2011 Jun;300(6):L898-909. doi: 10.1152/ajplung.00409.2010. Epub 2011 Apr 1. Am J Physiol Lung Cell Mol Physiol. 2011. PMID: 21460122 Free PMC article.
-
Short telomeres in pulmonary fibrosis: from genetics to clinical significance.Pneumologia. 2015 Jan-Mar;64(1):8, 11-3. Pneumologia. 2015. PMID: 26016050 Review.
-
The Role of Telomerase and Telomeres in Interstitial Lung Diseases: From Molecules to Clinical Implications.Int J Mol Sci. 2019 Jun 19;20(12):2996. doi: 10.3390/ijms20122996. Int J Mol Sci. 2019. PMID: 31248154 Free PMC article. Review.
Cited by
-
Telomerase deficiency does not alter bleomycin-induced fibrosis in mice.Exp Lung Res. 2012 Apr;38(3):124-34. doi: 10.3109/01902148.2012.658148. Exp Lung Res. 2012. PMID: 22394286 Free PMC article.
-
SIRT1 as a therapeutic target in inflammaging of the pulmonary disease.Prev Med. 2012 May;54 Suppl(Suppl):S20-8. doi: 10.1016/j.ypmed.2011.11.014. Epub 2011 Dec 8. Prev Med. 2012. PMID: 22178470 Free PMC article. Review.
-
WNT Signalling in Lung Physiology and Pathology.Handb Exp Pharmacol. 2021;269:305-336. doi: 10.1007/164_2021_521. Handb Exp Pharmacol. 2021. PMID: 34463851 Free PMC article.
-
Mechanisms of fibrosis: therapeutic translation for fibrotic disease.Nat Med. 2012 Jul 6;18(7):1028-40. doi: 10.1038/nm.2807. Nat Med. 2012. PMID: 22772564 Free PMC article. Review.
-
Pulmonary Alveolar Stem Cell Senescence, Apoptosis, and Differentiation by p53-Dependent and -Independent Mechanisms in Telomerase-Deficient Mice.Cells. 2021 Oct 26;10(11):2892. doi: 10.3390/cells10112892. Cells. 2021. PMID: 34831112 Free PMC article.
References
-
- Ahmed S, Passos JF, Birket MJ, Beckmann T, Brings S, Peters H, Birch-Machin MA, von Zglinicki T, Saretzki G. Telomerase does not counteract telomere shortening but protects mitochondrial function under oxidative stress. J Cell Sci 121: 1046–1053, 2008. - PubMed
-
- Albertine KH, Plopper CG. DNA oxidation or apoptosis: will the real culprit of DNA damage in hyperoxic lung injury please stand up? Am J Respir Cell Mol Biol 26: 381–383, 2002. - PubMed
-
- Armanios MY, Chen JJ, Cogan JD, Alder JK, Ingersoll RG, Markin C, Lawson WE, Xie M, Vulto I, Phillips JA 3rd, Lansdorp PM, Greider CW, Loyd JE. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med 356: 1317–1326, 2007. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous