

Calcineurin inhibitors modulate CXCR3 splice variant expression and mediate renal cancer progression
- PMID: 18832436
- PMCID: PMC2588111
- DOI: 10.1681/ASN.2008040394
Calcineurin inhibitors modulate CXCR3 splice variant expression and mediate renal cancer progression
Abstract
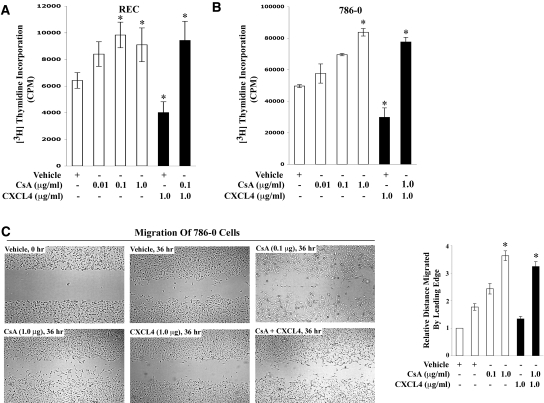
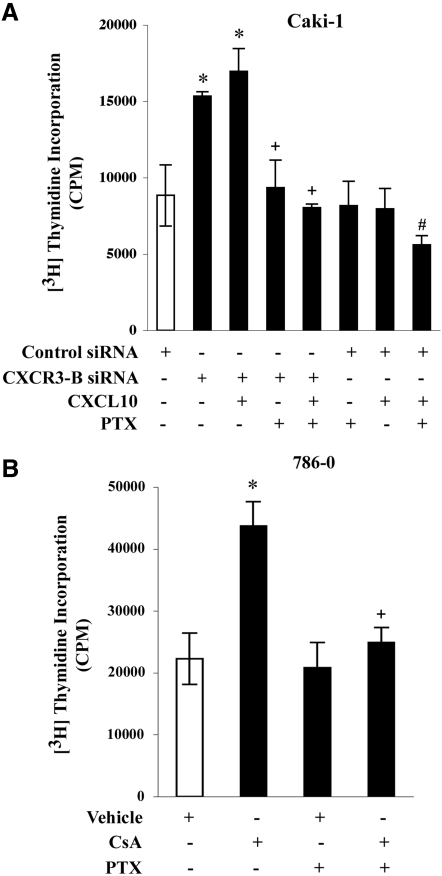
Calcineurin inhibitors (CNI) are used to prevent inflammatory diseases and allograft rejection. However, little is known about the mechanism(s) underlying their ability to promote the development and recurrence of cancer. Recent studies suggested that the chemokine receptor CXCR3 may play important roles in tumorigenesis. CXCR3 has two splice variants with opposite functions: CXCR3-A promotes cell proliferation, and CXCR3-B inhibits cell growth. Here, we explored the effects of CNI on the expression and function of CXCR3 splice variants. Compared with normal renal tissues and renal epithelial cells, human renal cancer tissues and renal cancer cell lines demonstrated higher expression of CXCR3-A and markedly lower expression of CXCR3-B. In human renal cancer cells (786-0 and Caki-1) and renal epithelial cells, CNI markedly downregulated the expression of CXCR3-B, whereas expression of CXCR3-A was unchanged. This CNI-mediated downregulation of CXCR3-B resulted in increased proliferation and migration of renal cancer cells; CNI-mediated cell proliferation involved signaling through G(i) proteins, perhaps via CXCR3-A. Finally, it was observed that CNI treatment increased the growth of human renal tumors in vivo, and the expression of CXCR3-B was significantly decreased in these tumors. In summary, these observations suggest that CNI may mediate the progression of human renal cancer by downregulating CXCR3-B and by promoting proliferative signals, likely through CXCR3-A. Targeting CXCR3 splice variants or the signaling pathways downstream of CXCR3 receptors may provide a therapeutic strategy for the prevention of CNI-mediated renal cancer progression.
Figures





Similar articles
-
CXCR3-B can mediate growth-inhibitory signals in human renal cancer cells by down-regulating the expression of heme oxygenase-1.J Biol Chem. 2010 Nov 19;285(47):36842-8. doi: 10.1074/jbc.M110.170324. Epub 2010 Sep 20. J Biol Chem. 2010. PMID: 20855888 Free PMC article.
-
Ras-induced modulation of CXCL10 and its receptor splice variant CXCR3-B in MDA-MB-435 and MCF-7 cells: relevance for the development of human breast cancer.Cancer Res. 2006 Oct 1;66(19):9509-18. doi: 10.1158/0008-5472.CAN-05-4345. Cancer Res. 2006. PMID: 17018607
-
Calcineurin inhibitors activate the proto-oncogene Ras and promote protumorigenic signals in renal cancer cells.Cancer Res. 2009 Dec 1;69(23):8902-9. doi: 10.1158/0008-5472.CAN-09-1404. Epub 2009 Nov 10. Cancer Res. 2009. PMID: 19903851 Free PMC article.
-
CXCR3 in carcinoma progression.Histol Histopathol. 2015 Jul;30(7):781-92. doi: 10.14670/HH-11-594. Epub 2015 Feb 9. Histol Histopathol. 2015. PMID: 25663474 Free PMC article. Review.
-
The CC and CXC chemokines: major regulators of tumor progression and the tumor microenvironment.Am J Physiol Cell Physiol. 2020 Mar 1;318(3):C542-C554. doi: 10.1152/ajpcell.00378.2019. Epub 2020 Jan 8. Am J Physiol Cell Physiol. 2020. PMID: 31913695 Free PMC article. Review.
Cited by
-
Differential expression profile of CXCR3 splicing variants is associated with thyroid neoplasia. Potential role in papillary thyroid carcinoma oncogenesis?Oncotarget. 2017 Dec 20;9(2):2445-2467. doi: 10.18632/oncotarget.23502. eCollection 2018 Jan 5. Oncotarget. 2017. PMID: 29416784 Free PMC article.
-
Achieving tolerance modifies cancer susceptibility profiles in liver transplant recipients.Cancer Med. 2023 Feb;12(4):5150-5157. doi: 10.1002/cam4.5271. Epub 2022 Oct 7. Cancer Med. 2023. PMID: 36205189 Free PMC article.
-
CXCR4 intracellular protein promotes drug resistance and tumorigenic potential by inversely regulating the expression of Death Receptor 5.Cell Death Dis. 2021 May 8;12(5):464. doi: 10.1038/s41419-021-03730-8. Cell Death Dis. 2021. PMID: 33966046 Free PMC article.
-
TNF-α augments CXCR2 and CXCR3 to promote progression of renal cell carcinoma.J Cell Mol Med. 2016 Nov;20(11):2020-2028. doi: 10.1111/jcmm.12890. Epub 2016 Jun 14. J Cell Mol Med. 2016. PMID: 27297979 Free PMC article.
-
A novel CXCR3-B chemokine receptor-induced growth-inhibitory signal in cancer cells is mediated through the regulation of Bach-1 protein and Nrf2 protein nuclear translocation.J Biol Chem. 2014 Feb 7;289(6):3126-37. doi: 10.1074/jbc.M113.508044. Epub 2013 Dec 23. J Biol Chem. 2014. PMID: 24366869 Free PMC article.
References
-
- Klee CB, Ren H, Wang X: Regulation of the calmodulin-stimulated protein phosphatase, calcineurin. J Biol Chem 273: 13367–13370, 1998 - PubMed
-
- Graef IA, Chen F, Chen L, Kuo A, Crabtree GR: Signals transduced by Ca(2+)/calcineurin and NFATc3/c4 pattern the developing vasculature. Cell 105: 863–875, 2001 - PubMed
-
- Crabtree GR, Olson EN: NFAT signaling: Choreographing the social lives of cells. Cell 109[Suppl]: S67–S79, 2002 - PubMed
-
- Hogan PG, Chen L, Nardone J, Rao A: Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev 17: 2205–2232, 2003 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
