

The unfoldomics decade: an update on intrinsically disordered proteins
- PMID: 18831774
- PMCID: PMC2559873
- DOI: 10.1186/1471-2164-9-S2-S1
The unfoldomics decade: an update on intrinsically disordered proteins
Abstract
Background: Our first predictor of protein disorder was published just over a decade ago in the Proceedings of the IEEE International Conference on Neural Networks (Romero P, Obradovic Z, Kissinger C, Villafranca JE, Dunker AK (1997) Identifying disordered regions in proteins from amino acid sequence. Proceedings of the IEEE International Conference on Neural Networks, 1: 90-95). By now more than twenty other laboratory groups have joined the efforts to improve the prediction of protein disorder. While the various prediction methodologies used for protein intrinsic disorder resemble those methodologies used for secondary structure prediction, the two types of structures are entirely different. For example, the two structural classes have very different dynamic properties, with the irregular secondary structure class being much less mobile than the disorder class. The prediction of secondary structure has been useful. On the other hand, the prediction of intrinsic disorder has been revolutionary, leading to major modifications of the more than 100 year-old views relating protein structure and function. Experimentalists have been providing evidence over many decades that some proteins lack fixed structure or are disordered (or unfolded) under physiological conditions. In addition, experimentalists are also showing that, for many proteins, their functions depend on the unstructured rather than structured state; such results are in marked contrast to the greater than hundred year old views such as the lock and key hypothesis. Despite extensive data on many important examples, including disease-associated proteins, the importance of disorder for protein function has been largely ignored. Indeed, to our knowledge, current biochemistry books don't present even one acknowledged example of a disorder-dependent function, even though some reports of disorder-dependent functions are more than 50 years old. The results from genome-wide predictions of intrinsic disorder and the results from other bioinformatics studies of intrinsic disorder are demanding attention for these proteins.
Results: Disorder prediction has been important for showing that the relatively few experimentally characterized examples are members of a very large collection of related disordered proteins that are wide-spread over all three domains of life. Many significant biological functions are now known to depend directly on, or are importantly associated with, the unfolded or partially folded state. Here our goal is to review the key discoveries and to weave these discoveries together to support novel approaches for understanding sequence-function relationships.
Conclusion: Intrinsically disordered protein is common across the three domains of life, but especially common among the eukaryotic proteomes. Signaling sequences and sites of posttranslational modifications are frequently, or very likely most often, located within regions of intrinsic disorder. Disorder-to-order transitions are coupled with the adoption of different structures with different partners. Also, the flexibility of intrinsic disorder helps different disordered regions to bind to a common binding site on a common partner. Such capacity for binding diversity plays important roles in both protein-protein interaction networks and likely also in gene regulation networks. Such disorder-based signaling is further modulated in multicellular eukaryotes by alternative splicing, for which such splicing events map to regions of disorder much more often than to regions of structure. Associating alternative splicing with disorder rather than structure alleviates theoretical and experimentally observed problems associated with the folding of different length, isomeric amino acid sequences. The combination of disorder and alternative splicing is proposed to provide a mechanism for easily "trying out" different signaling pathways, thereby providing the mechanism for generating signaling diversity and enabling the evolution of cell differentiation and multicellularity. Finally, several recent small molecules of interest as potential drugs have been shown to act by blocking protein-protein interactions based on intrinsic disorder of one of the partners. Study of these examples has led to a new approach for drug discovery, and bioinformatics analysis of the human proteome suggests that various disease-associated proteins are very rich in such disorder-based drug discovery targets.
Figures
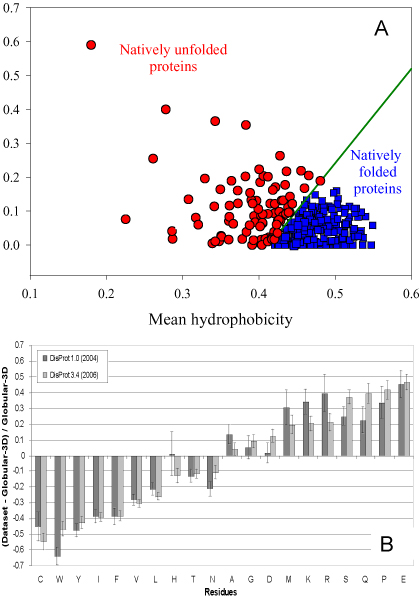
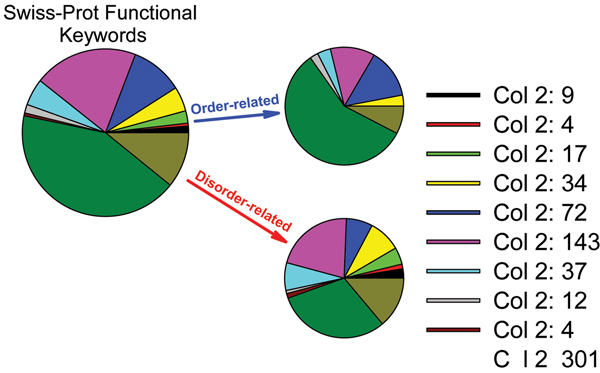
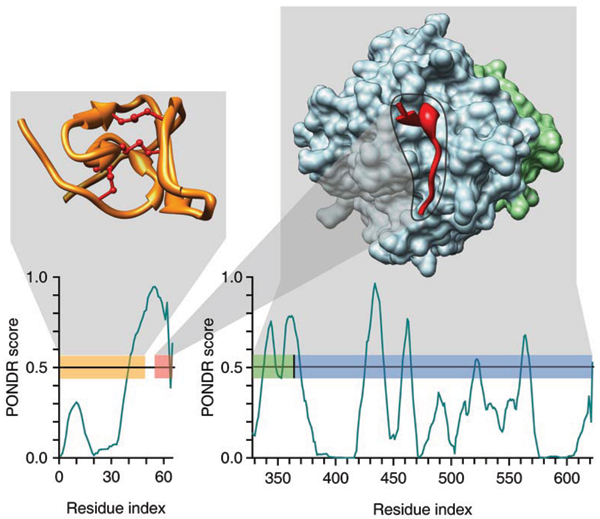

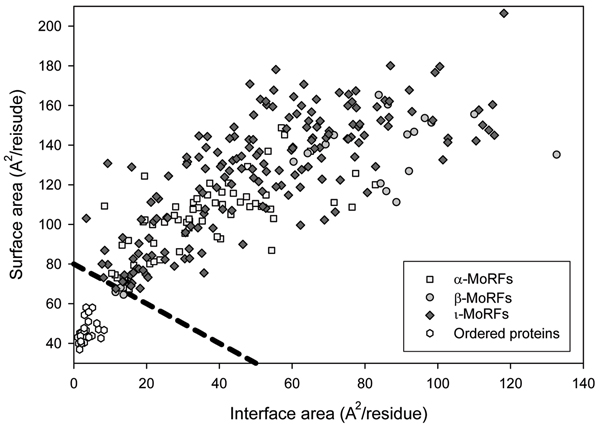
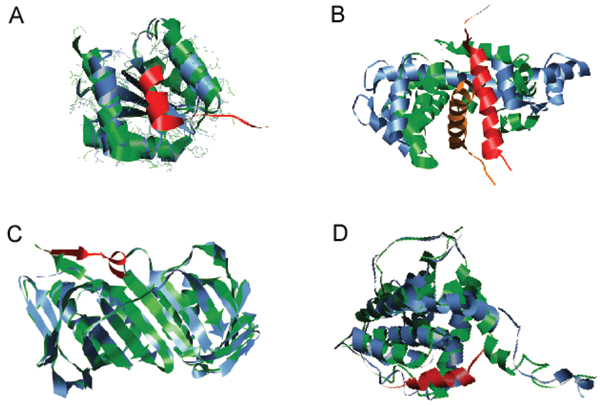
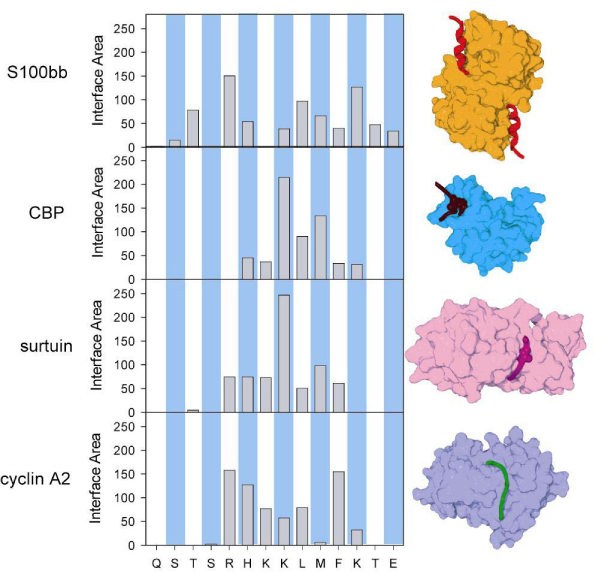
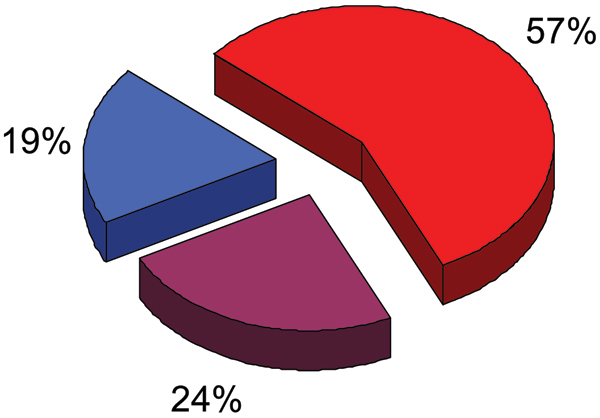
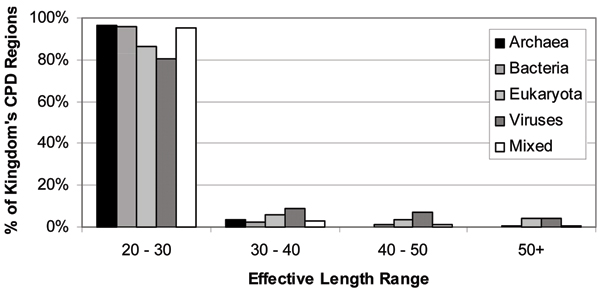
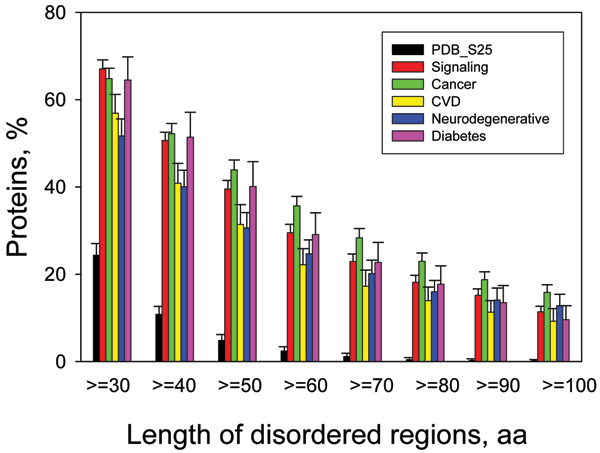
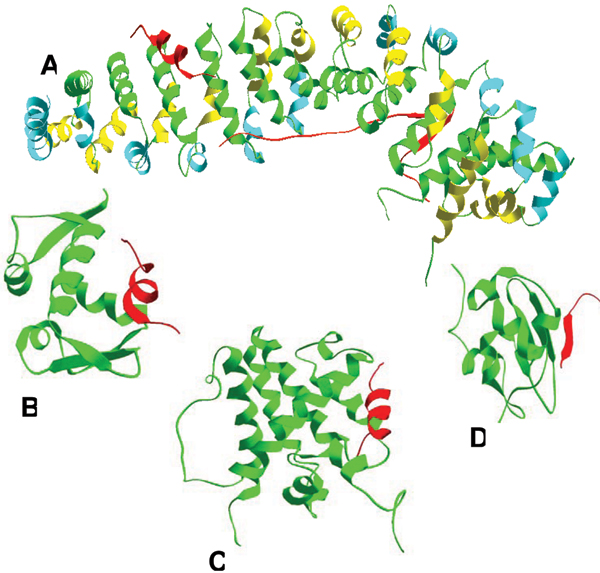
Similar articles
-
Macromolecular crowding: chemistry and physics meet biology (Ascona, Switzerland, 10-14 June 2012).Phys Biol. 2013 Aug;10(4):040301. doi: 10.1088/1478-3975/10/4/040301. Epub 2013 Aug 2. Phys Biol. 2013. PMID: 23912807
-
Functional anthology of intrinsic disorder. 3. Ligands, post-translational modifications, and diseases associated with intrinsically disordered proteins.J Proteome Res. 2007 May;6(5):1917-32. doi: 10.1021/pr060394e. Epub 2007 Mar 29. J Proteome Res. 2007. PMID: 17391016 Free PMC article.
-
Unfoldomics of human diseases: linking protein intrinsic disorder with diseases.BMC Genomics. 2009 Jul 7;10 Suppl 1(Suppl 1):S7. doi: 10.1186/1471-2164-10-S1-S7. BMC Genomics. 2009. PMID: 19594884 Free PMC article.
-
Function and structure of inherently disordered proteins.Curr Opin Struct Biol. 2008 Dec;18(6):756-64. doi: 10.1016/j.sbi.2008.10.002. Epub 2008 Nov 17. Curr Opin Struct Biol. 2008. PMID: 18952168 Review.
-
Natively disordered proteins: functions and predictions.Appl Bioinformatics. 2004;3(2-3):105-13. doi: 10.2165/00822942-200403020-00005. Appl Bioinformatics. 2004. PMID: 15693736 Review.
Cited by
-
Molecular phylogeny of OVOL genes illustrates a conserved C2H2 zinc finger domain coupled by hypervariable unstructured regions.PLoS One. 2012;7(6):e39399. doi: 10.1371/journal.pone.0039399. Epub 2012 Jun 21. PLoS One. 2012. PMID: 22737237 Free PMC article.
-
Computational and statistical analyses of amino acid usage and physico-chemical properties of the twelve late embryogenesis abundant protein classes.PLoS One. 2012;7(5):e36968. doi: 10.1371/journal.pone.0036968. Epub 2012 May 16. PLoS One. 2012. PMID: 22615859 Free PMC article.
-
Connecting autophagy: AMBRA1 and its network of regulation.Mol Cell Oncol. 2015 Feb 24;2(1):e970059. doi: 10.4161/23723548.2014.970059. eCollection 2015 Jan-Mar. Mol Cell Oncol. 2015. PMID: 27308402 Free PMC article. Review.
-
The human selenoprotein VCP-interacting membrane protein (VIMP) is non-globular and harbors a reductase function in an intrinsically disordered region.J Biol Chem. 2012 Jul 27;287(31):26388-99. doi: 10.1074/jbc.M112.346775. Epub 2012 Jun 14. J Biol Chem. 2012. PMID: 22700979 Free PMC article.
-
Multicolor single-molecule FRET to explore protein folding and binding.Mol Biosyst. 2010 Sep;6(9):1540-7. doi: 10.1039/c003024d. Epub 2010 Jul 2. Mol Biosyst. 2010. PMID: 20601974 Free PMC article. Review.
References
-
- Landsteiner K. The specificity of serological reactions. Baltimore: C. C. Thomas; 1936.
-
- Pauling L. A Theory of the Structure and Process of Formation of Antibodies. J Am Chem Soc. 1940;62:2643–2657.
-
- Dunker AK, Garner E, Guilliot S, Romero P, Albrecht K, Hart J, Obradovic Z, Kissinger C, Villafranca JE. Protein disorder and the evolution of molecular recognition: theory, predictions and observations. Pac Symp Biocomput. 1998:473–484. - PubMed
-
- Karush F. Heterogeneity of the binding sites of bovine serum albumin. J Am Chem Soc. 1950;72:2705–2713.
-
- Spolar RS, Record MT., Jr Coupling of local folding to site-specific binding of proteins to DNA. Science. 1994;263:777–784. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
