

A feature-based approach to modeling protein-DNA interactions
- PMID: 18725950
- PMCID: PMC2516605
- DOI: 10.1371/journal.pcbi.1000154
A feature-based approach to modeling protein-DNA interactions
Abstract
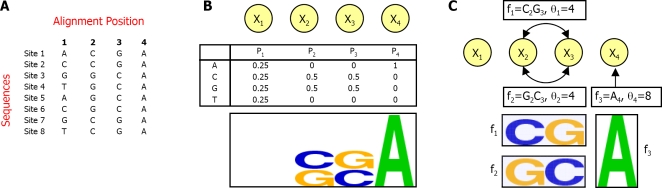
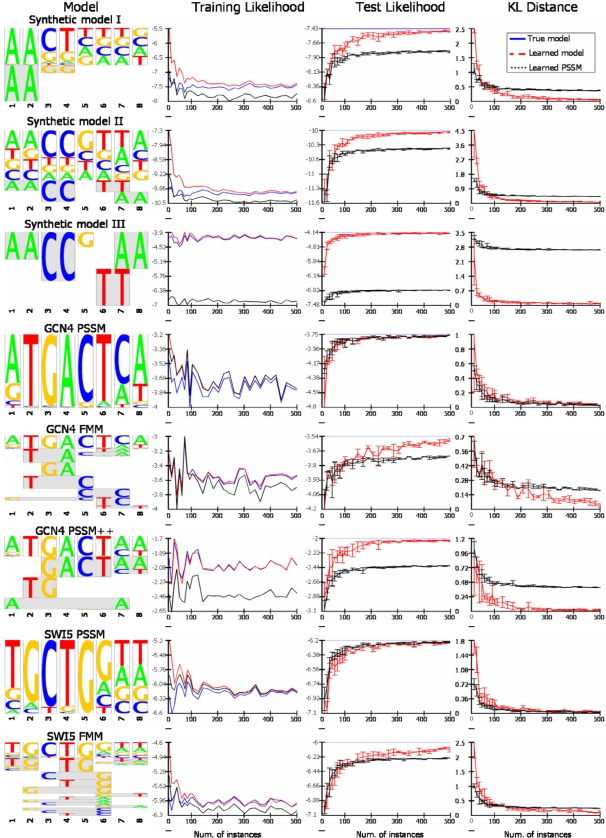
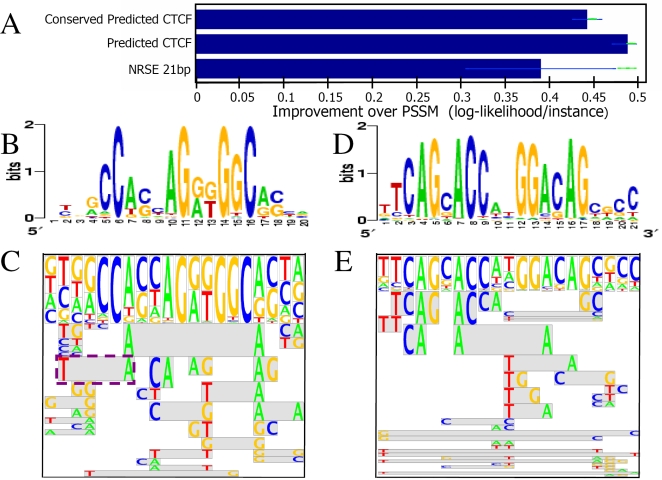
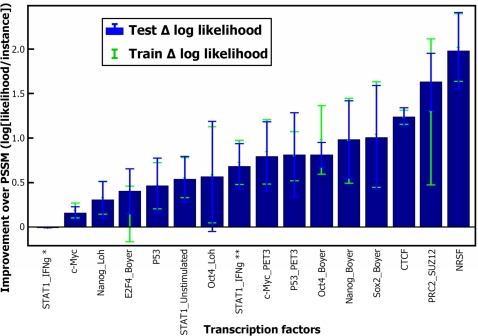
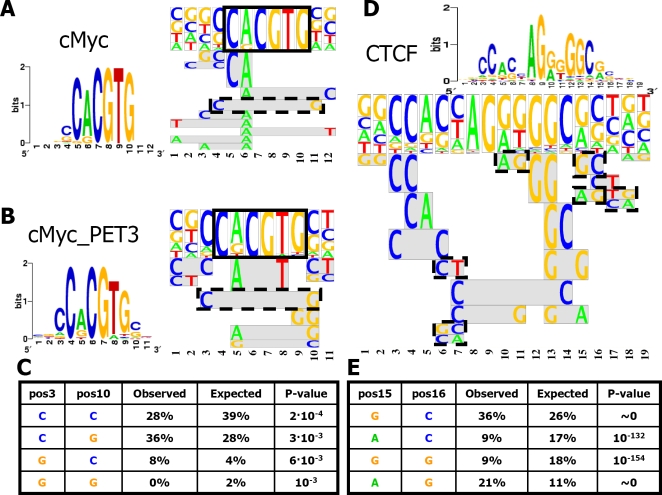
Transcription factor (TF) binding to its DNA target site is a fundamental regulatory interaction. The most common model used to represent TF binding specificities is a position specific scoring matrix (PSSM), which assumes independence between binding positions. However, in many cases, this simplifying assumption does not hold. Here, we present feature motif models (FMMs), a novel probabilistic method for modeling TF-DNA interactions, based on log-linear models. Our approach uses sequence features to represent TF binding specificities, where each feature may span multiple positions. We develop the mathematical formulation of our model and devise an algorithm for learning its structural features from binding site data. We also developed a discriminative motif finder, which discovers de novo FMMs that are enriched in target sets of sequences compared to background sets. We evaluate our approach on synthetic data and on the widely used TF chromatin immunoprecipitation (ChIP) dataset of Harbison et al. We then apply our algorithm to high-throughput TF ChIP data from mouse and human, reveal sequence features that are present in the binding specificities of mouse and human TFs, and show that FMMs explain TF binding significantly better than PSSMs. Our FMM learning and motif finder software are available at http://genie.weizmann.ac.il/.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures


Similar articles
-
High resolution models of transcription factor-DNA affinities improve in vitro and in vivo binding predictions.PLoS Comput Biol. 2010 Sep 9;6(9):e1000916. doi: 10.1371/journal.pcbi.1000916. PLoS Comput Biol. 2010. PMID: 20838582 Free PMC article.
-
A biophysical model for analysis of transcription factor interaction and binding site arrangement from genome-wide binding data.PLoS One. 2009 Dec 1;4(12):e8155. doi: 10.1371/journal.pone.0008155. PLoS One. 2009. PMID: 19956545 Free PMC article.
-
Identification of context-dependent motifs by contrasting ChIP binding data.Bioinformatics. 2010 Nov 15;26(22):2826-32. doi: 10.1093/bioinformatics/btq546. Epub 2010 Sep 23. Bioinformatics. 2010. PMID: 20870645 Free PMC article.
-
Protein binding microarrays for the characterization of DNA-protein interactions.Adv Biochem Eng Biotechnol. 2007;104:65-85. doi: 10.1007/10_025. Adv Biochem Eng Biotechnol. 2007. PMID: 17290819 Free PMC article. Review.
-
Analysis of sequence specificities of DNA-binding proteins with protein binding microarrays.Methods Enzymol. 2006;410:279-99. doi: 10.1016/S0076-6879(06)10013-0. Methods Enzymol. 2006. PMID: 16938556 Free PMC article. Review.
Cited by
-
Protein-DNA binding in high-resolution.Crit Rev Biochem Mol Biol. 2015;50(4):269-83. doi: 10.3109/10409238.2015.1051505. Epub 2015 Jun 3. Crit Rev Biochem Mol Biol. 2015. PMID: 26038153 Free PMC article. Review.
-
Experimental strategies for studying transcription factor-DNA binding specificities.Brief Funct Genomics. 2010 Dec;9(5-6):362-73. doi: 10.1093/bfgp/elq023. Epub 2010 Sep 23. Brief Funct Genomics. 2010. PMID: 20864494 Free PMC article. Review.
-
Metamotifs--a generative model for building families of nucleotide position weight matrices.BMC Bioinformatics. 2010 Jun 25;11:348. doi: 10.1186/1471-2105-11-348. BMC Bioinformatics. 2010. PMID: 20579334 Free PMC article.
-
Statistical Analyses of Next Generation Sequence Data: A Partial Overview.J Proteomics Bioinform. 2010 Jun 1;3(6):183-190. doi: 10.4172/jpb.1000138. J Proteomics Bioinform. 2010. PMID: 21113236 Free PMC article.
-
Fast matching of transcription factor motifs using generalized position weight matrix models.J Comput Biol. 2013 Sep;20(9):621-30. doi: 10.1089/cmb.2012.0289. Epub 2013 Aug 6. J Comput Biol. 2013. PMID: 23919388 Free PMC article.
References
-
- Elnitski L, Jin VX, Farnham PJ, Jones SJ. Locating mammalian transcription factor binding sites: a survey of computational and experimental techniques. Genome Res. 2006;16:1455–1464. - PubMed
-
- Bulyk ML, Gentalen E, Lockhart DJ, Church GM. Quantifying DNA–protein interactions by double-stranded DNA arrays. Nat Biotechnol. 1999;17:573–577. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
