

The aryl hydrocarbon receptor attenuates tobacco smoke-induced cyclooxygenase-2 and prostaglandin production in lung fibroblasts through regulation of the NF-kappaB family member RelB
- PMID: 18697742
- PMCID: PMC2570856
- DOI: 10.1074/jbc.M800685200
The aryl hydrocarbon receptor attenuates tobacco smoke-induced cyclooxygenase-2 and prostaglandin production in lung fibroblasts through regulation of the NF-kappaB family member RelB
Abstract
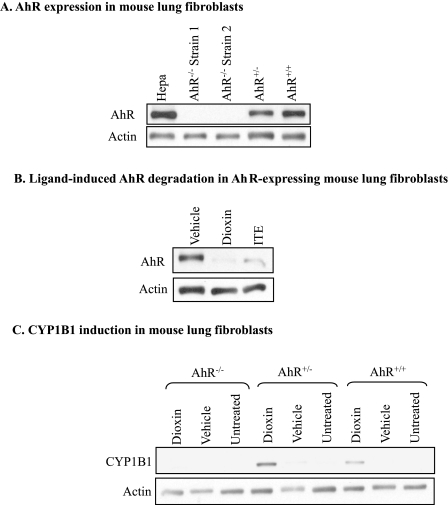
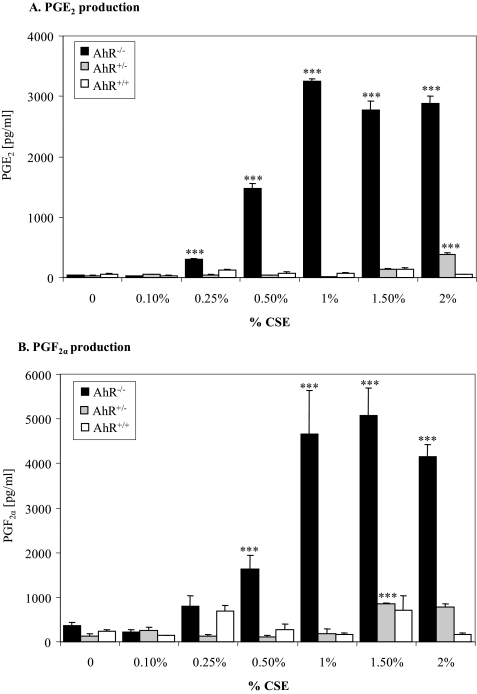
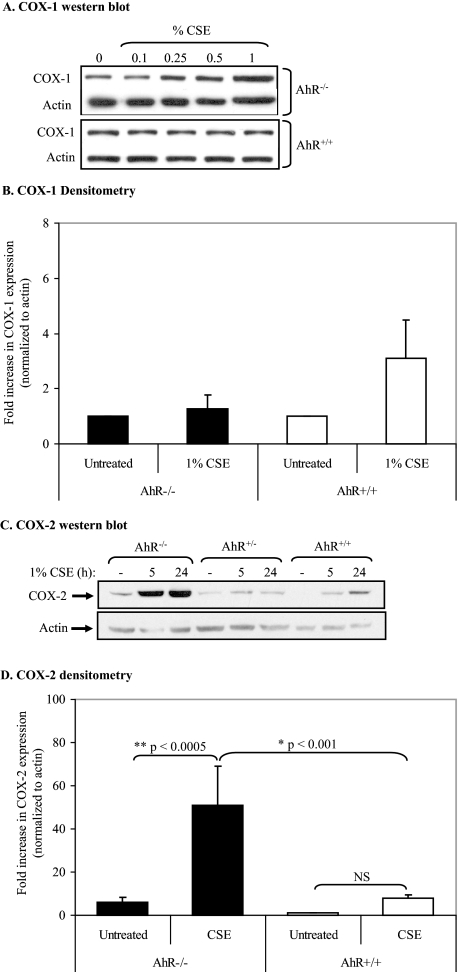
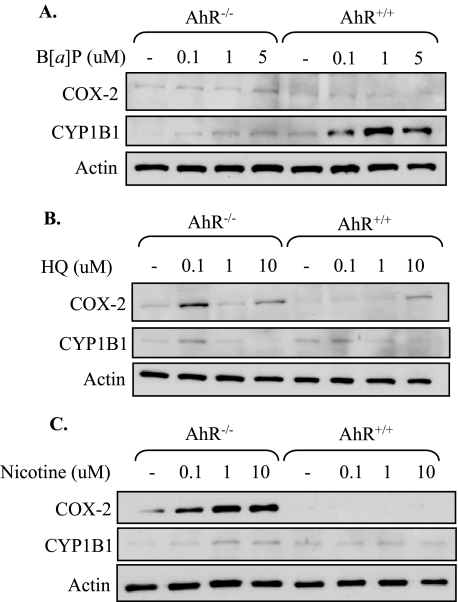
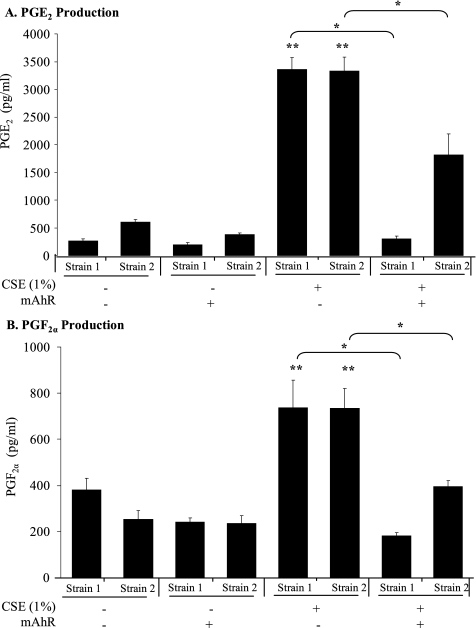
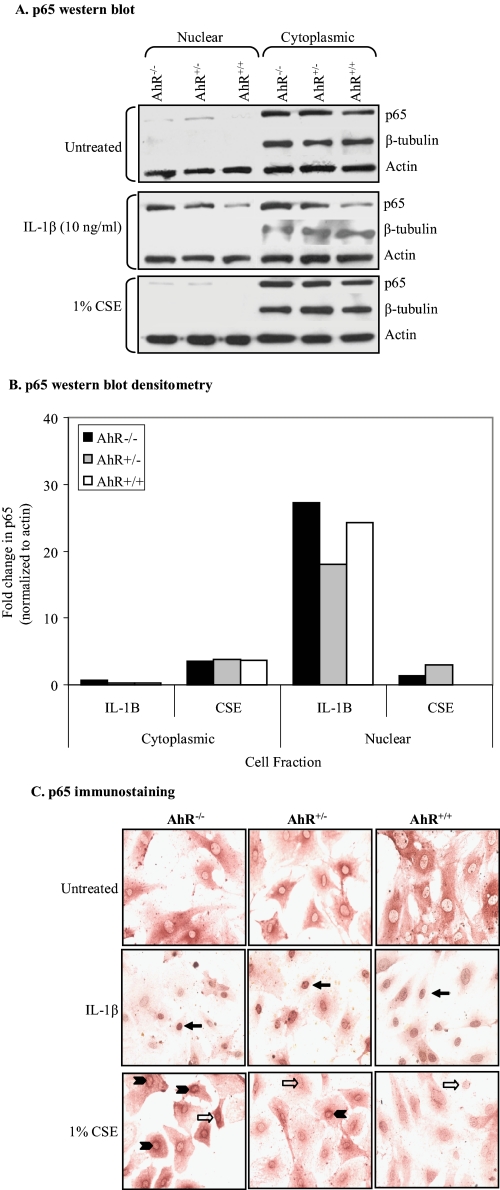
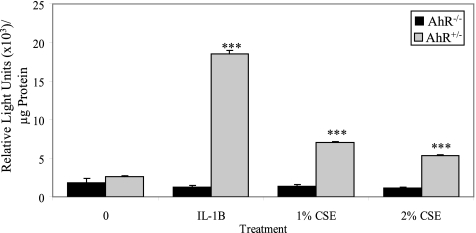
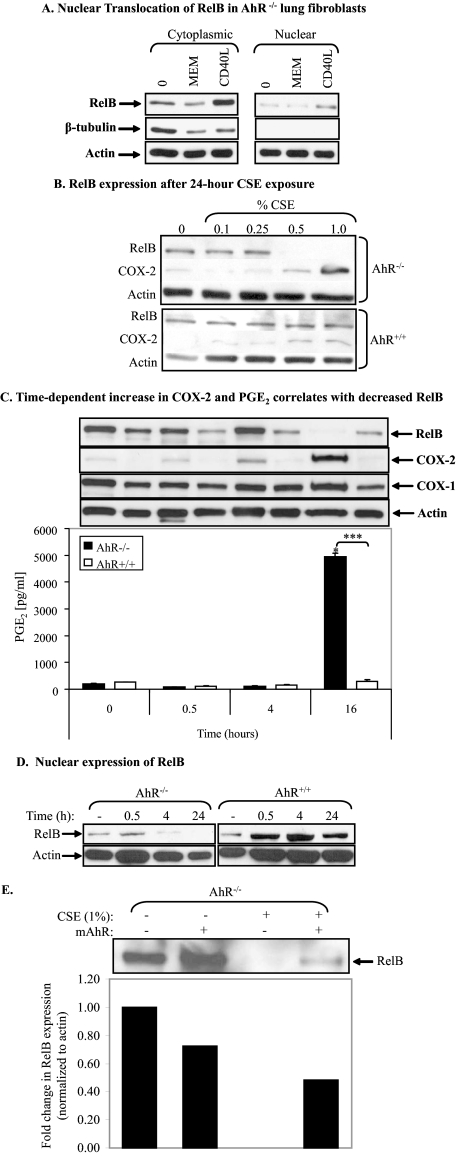
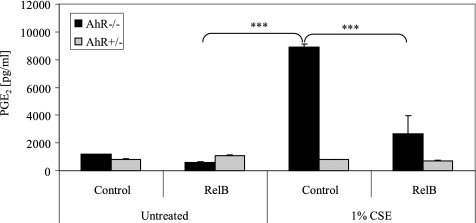
Diseases such as chronic obstructive pulmonary disease and lung cancer caused by cigarette smoke affect millions of people worldwide. The aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor that influences responses to certain environmental pollutants such as tobacco smoke. However, the physiological function(s) of the AhR is unknown. Herein we propose that the physiologic role of the AhR is to limit inflammation. We show that lung fibroblasts from AhR(-/-) mice produce a heightened inflammatory response to cigarette smoke, typified by increased levels of cyclooxygenase-2 (COX-2) and prostaglandins (PGs), when compared with wild type (AhR(+/+)) fibroblasts. This response was dependent on AhR expression as transient transfection of an AhR expression plasmid into AhR(-/-) fibroblasts significantly attenuated the smoke-induced COX-2 and PG production, confirming the anti-inflammatory role of the AhR. The AhR can interact with NF-kappaB. However, the heightened inflammatory response observed in AhR(-/-) fibroblasts was not the result of NF-kappaB (p50/p65) activation. Instead it was coupled with a loss of the NF-kappaB family member RelB in AhR(-/-) fibroblasts. Taken together, these studies provide compelling evidence that AhR expression limits proinflammatory COX-2 and PG production by maintaining RelB expression. The association between RelB and AhR may represent a new therapeutic and more selective target with which to combat inflammation-associated diseases.
Figures


Similar articles
-
The aryl hydrocarbon receptor is a regulator of cigarette smoke induction of the cyclooxygenase and prostaglandin pathways in human lung fibroblasts.Am J Physiol Lung Cell Mol Physiol. 2005 Sep;289(3):L391-9. doi: 10.1152/ajplung.00062.2005. Epub 2005 Apr 29. Am J Physiol Lung Cell Mol Physiol. 2005. PMID: 15863442
-
Depressing time: Waiting, melancholia, and the psychoanalytic practice of care.In: Kirtsoglou E, Simpson B, editors. The Time of Anthropology: Studies of Contemporary Chronopolitics. Abingdon: Routledge; 2020. Chapter 5. In: Kirtsoglou E, Simpson B, editors. The Time of Anthropology: Studies of Contemporary Chronopolitics. Abingdon: Routledge; 2020. Chapter 5. PMID: 36137063 Free Books & Documents. Review.
-
Qualitative evidence synthesis informing our understanding of people's perceptions and experiences of targeted digital communication.Cochrane Database Syst Rev. 2019 Oct 23;10(10):ED000141. doi: 10.1002/14651858.ED000141. Cochrane Database Syst Rev. 2019. PMID: 31643081 Free PMC article.
-
Comparison of Two Modern Survival Prediction Tools, SORG-MLA and METSSS, in Patients With Symptomatic Long-bone Metastases Who Underwent Local Treatment With Surgery Followed by Radiotherapy and With Radiotherapy Alone.Clin Orthop Relat Res. 2024 Dec 1;482(12):2193-2208. doi: 10.1097/CORR.0000000000003185. Epub 2024 Jul 23. Clin Orthop Relat Res. 2024. PMID: 39051924
-
A consideration of the role of gas/particle partitioning in the deposition of nicotine and other tobacco smoke compounds in the respiratory tract.Chem Res Toxicol. 2001 Nov;14(11):1465-81. doi: 10.1021/tx0100901. Chem Res Toxicol. 2001. PMID: 11712903 Review.
Cited by
-
Breaking the mold: transcription factors in the anucleate platelet and platelet-derived microparticles.Front Immunol. 2015 Feb 13;6:48. doi: 10.3389/fimmu.2015.00048. eCollection 2015. Front Immunol. 2015. PMID: 25762994 Free PMC article. Review.
-
Exposure to Heptachlorodibenzo-p-dioxin (HpCDD) Regulates microRNA Expression in Human Lung Fibroblasts.J Occup Environ Med. 2019 Dec;61 Suppl 12(Suppl 12):S82-S89. doi: 10.1097/JOM.0000000000001691. J Occup Environ Med. 2019. PMID: 31800454 Free PMC article.
-
Animal models of drug-induced pulmonary fibrosis: an overview of molecular mechanisms and characteristics.Cell Biol Toxicol. 2022 Oct;38(5):699-723. doi: 10.1007/s10565-021-09676-z. Epub 2021 Nov 5. Cell Biol Toxicol. 2022. PMID: 34741237 Review.
-
Adverse effects of electronic cigarettes on the disease-naive oral microbiome.Sci Adv. 2020 May 27;6(22):eaaz0108. doi: 10.1126/sciadv.aaz0108. eCollection 2020 May. Sci Adv. 2020. PMID: 32518820 Free PMC article.
-
Endogenous ligands of the aryl hydrocarbon receptor regulate lung dendritic cell function.Immunology. 2016 Jan;147(1):41-54. doi: 10.1111/imm.12540. Epub 2015 Nov 10. Immunology. 2016. PMID: 26555456 Free PMC article.
References
-
- Hida, T., Yatabe, Y., Achiwa, H., Muramatsu, H., Kozaki, K., Nakamura, S., Ogawa, M., Mitsudomi, T., Sugiura, T., and Takahashi, T. (1998) Cancer Res. 58 3761–3764 - PubMed
-
- Xaubet, A., Roca-Ferrer, J., Pujols, L., Ramirez, J., Mullol, J., Marin-Arguedas, A., Torrego, A., Gimferrer, J. M., and Picado, C. (2004) Sarcoidosis Vasc. Diffuse Lung Dis. 21 35–42 - PubMed
-
- Kamiyama, M., Pozzi, A., Yang, L., DeBusk, L. M., Breyer, R. M., and Lin, P. C. (2006) Oncogene 25 7019–7028 - PubMed
-
- Subbaramaiah, K., and Dannenberg, A. J. (2003) Trends Pharmacol. Sci. 24 96–102 - PubMed
-
- Smith, W. L., DeWitt, D. L., and Garavito, R. M. (2000) Annu. Rev. Biochem. 69 145–182 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
