

Cotranslational folding promotes beta-helix formation and avoids aggregation in vivo
- PMID: 18674543
- PMCID: PMC2597226
- DOI: 10.1016/j.jmb.2008.07.035
Cotranslational folding promotes beta-helix formation and avoids aggregation in vivo
Abstract
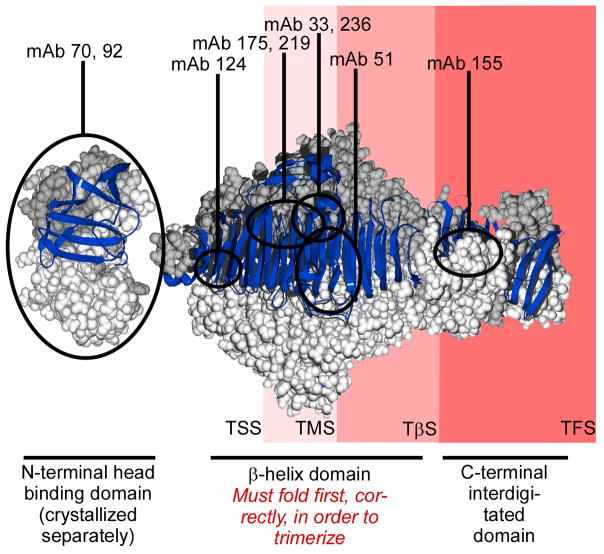
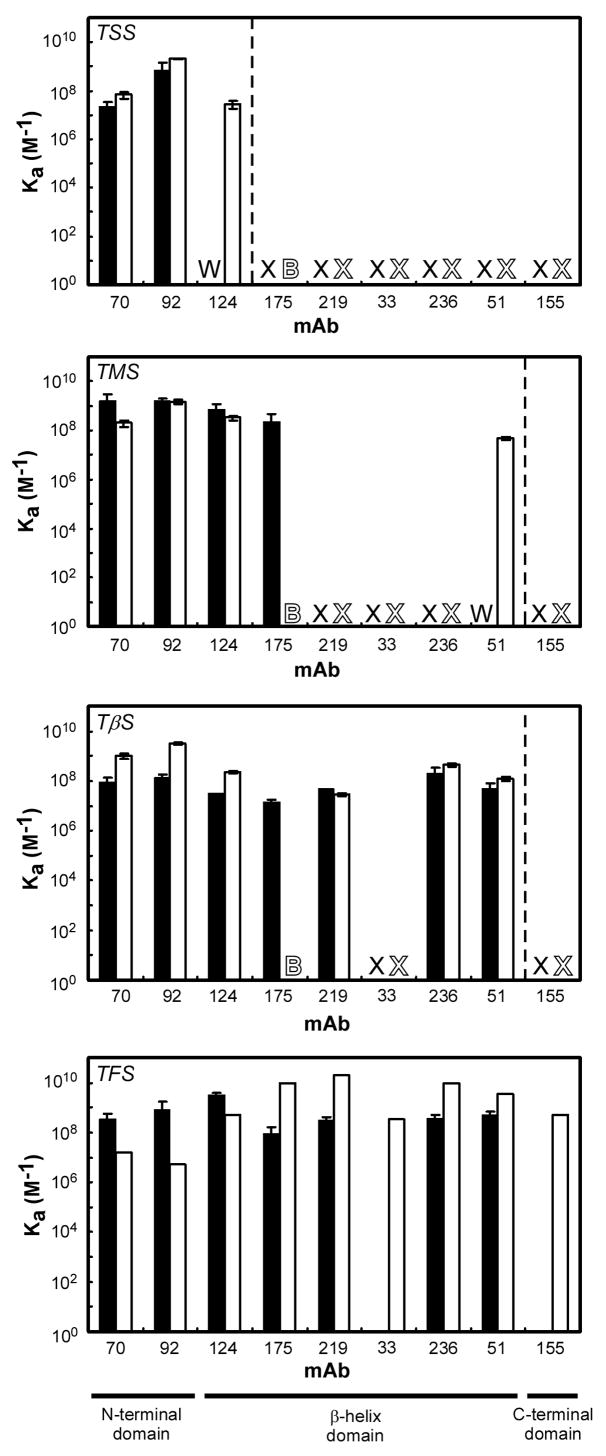
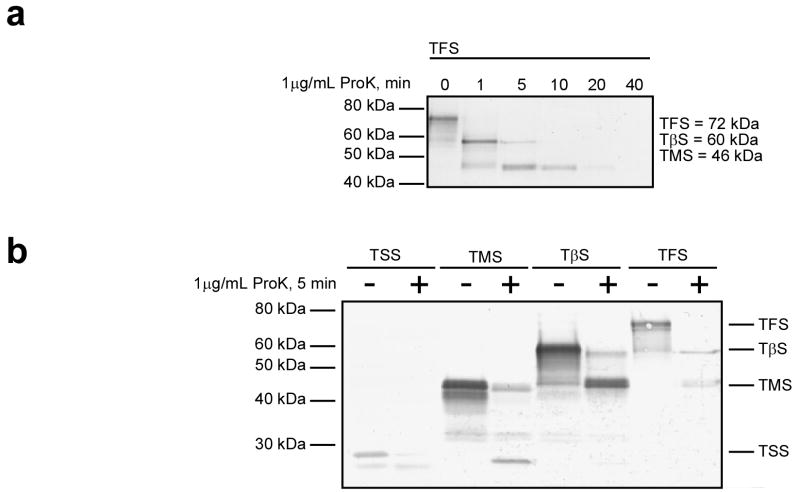
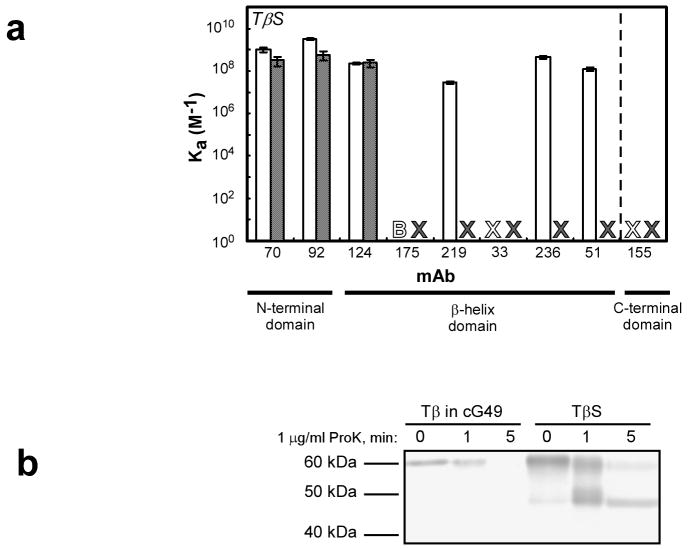
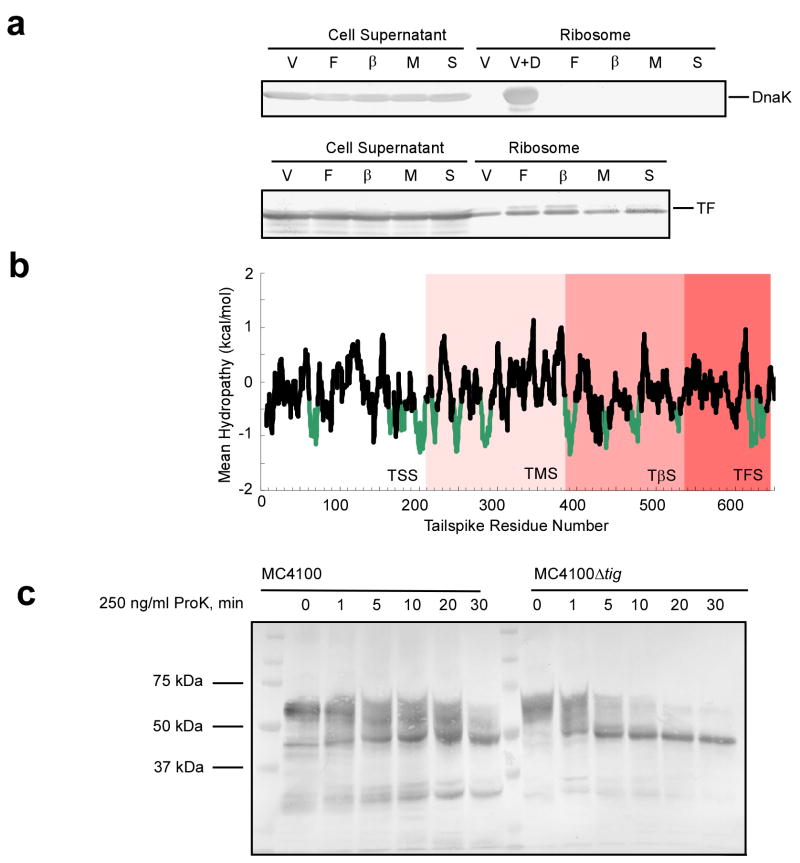
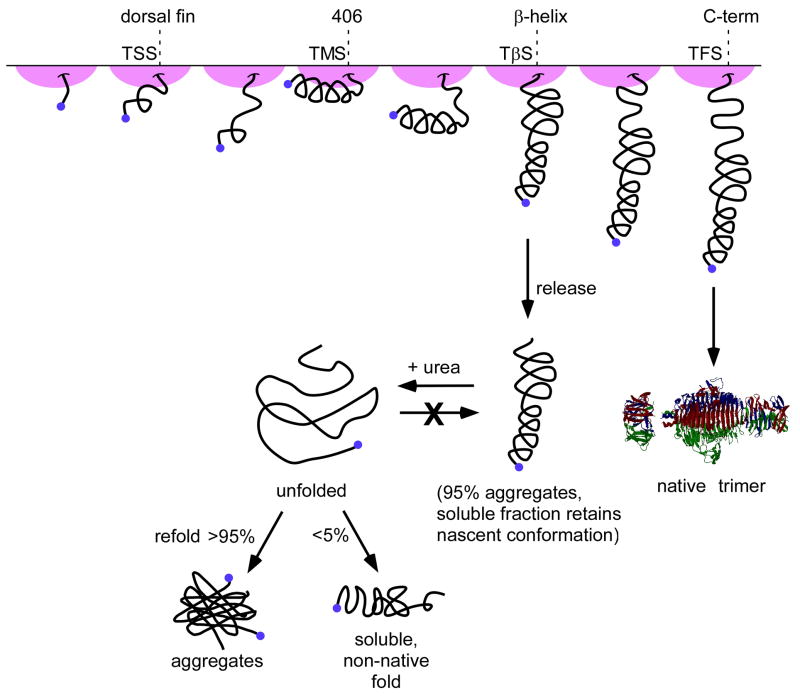
Newly synthesized proteins must form their native structures in the crowded environment of the cell, while avoiding non-native conformations that can lead to aggregation. Yet, remarkably little is known about the progressive folding of polypeptide chains during chain synthesis by the ribosome or of the influence of this folding environment on productive folding in vivo. P22 tailspike is a homotrimeric protein that is prone to aggregation via misfolding of its central beta-helix domain in vitro. We have produced stalled ribosome:tailspike nascent chain complexes of four fixed lengths in vivo, in order to assess cotranslational folding of newly synthesized tailspike chains as a function of chain length. Partially synthesized, ribosome-bound nascent tailspike chains populate stable conformations with some native-state structural features even prior to the appearance of the entire beta-helix domain, regardless of the presence of the chaperone trigger factor, yet these conformations are distinct from the conformations of released, refolded tailspike truncations. These results suggest that organization of the aggregation-prone beta-helix domain occurs cotranslationally, prior to chain release, to a conformation that is distinct from the accessible energy minimum conformation for the truncated free chain in solution.
Figures
Similar articles
-
A newly synthesized, ribosome-bound polypeptide chain adopts conformations dissimilar from early in vitro refolding intermediates.J Biol Chem. 2001 Jul 6;276(27):25411-20. doi: 10.1074/jbc.M008490200. Epub 2001 Apr 23. J Biol Chem. 2001. PMID: 11319217
-
A reversibly unfolding fragment of P22 tailspike protein with native structure: the isolated beta-helix domain.Biochemistry. 1998 Jun 23;37(25):9160-8. doi: 10.1021/bi980190e. Biochemistry. 1998. PMID: 9636063
-
Plasticity and steric strain in a parallel beta-helix: rational mutations in the P22 tailspike protein.Proteins. 2000 Apr 1;39(1):89-101. Proteins. 2000. PMID: 10737931
-
There's a right way and a wrong way: in vivo and in vitro folding, misfolding and subunit assembly of the P22 tailspike.Structure. 1999 Jun 15;7(6):R131-9. doi: 10.1016/s0969-2126(99)80078-1. Structure. 1999. PMID: 10404587 Review.
-
Phage tailspike protein. A fishy tale of protein folding.Curr Biol. 1994 Nov 1;4(11):1026-9. doi: 10.1016/s0960-9822(00)00234-7. Curr Biol. 1994. PMID: 7874487 Review.
Cited by
-
Folding the proteome.Trends Biochem Sci. 2013 Jul;38(7):337-44. doi: 10.1016/j.tibs.2013.05.001. Epub 2013 Jun 11. Trends Biochem Sci. 2013. PMID: 23764454 Free PMC article. Review.
-
A strategy for co-translational folding studies of ribosome-bound nascent chain complexes using NMR spectroscopy.Nat Protoc. 2016 Aug;11(8):1492-507. doi: 10.1038/nprot.2016.101. Epub 2016 Jul 28. Nat Protoc. 2016. PMID: 27466710
-
Effect of Protein Structure on Evolution of Cotranslational Folding.Biophys J. 2020 Sep 15;119(6):1123-1134. doi: 10.1016/j.bpj.2020.06.037. Epub 2020 Aug 12. Biophys J. 2020. PMID: 32857962 Free PMC article.
-
The ribosome modulates nascent protein folding.Science. 2011 Dec 23;334(6063):1723-7. doi: 10.1126/science.1209740. Science. 2011. PMID: 22194581 Free PMC article.
-
The ribosome in action: Tuning of translational efficiency and protein folding.Protein Sci. 2016 Aug;25(8):1390-406. doi: 10.1002/pro.2950. Epub 2016 Jun 8. Protein Sci. 2016. PMID: 27198711 Free PMC article. Review.
References
-
- Ellis RJ. Macromolecular crowding: Obvious but underappreciated. Trends Biochem Sci. 2001;26:597–604. - PubMed
-
- Frydman J, Erdjument-Bromage H, Tempst P, Hartl FU. Co-translational domain folding as the structural basis for the rapid de novo folding of firefly luciferase. Nat Struct Biol. 1999;6:697–705. - PubMed
-
- Fedorov AN, Baldwin TO. Process of biosynthetic protein folding determines the rapid formation of native structure. J Mol Biol. 1999;294:579–586. - PubMed
-
- Netzer WJ, Hartl FU. Recombination of protein domains facilitated by co-translational folding in eukaryotes. Nature. 1997;388:343–349. - PubMed
-
- Nicola AV, Chen W, Helenius A. Co-translational folding of an alphavirus capsid protein in the cytosol of living cells. Nat Cell Biol. 1999;1:341–345. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
