

Accelerating and focusing protein-protein docking correlations using multi-dimensional rotational FFT generating functions
- PMID: 18591193
- PMCID: PMC2732220
- DOI: 10.1093/bioinformatics/btn334
Accelerating and focusing protein-protein docking correlations using multi-dimensional rotational FFT generating functions
Abstract
Motivation: Predicting how proteins interact at the molecular level is a computationally intensive task. Many protein docking algorithms begin by using fast Fourier transform (FFT) correlation techniques to find putative rigid body docking orientations. Most such approaches use 3D Cartesian grids and are therefore limited to computing three dimensional (3D) translational correlations. However, translational FFTs can speed up the calculation in only three of the six rigid body degrees of freedom, and they cannot easily incorporate prior knowledge about a complex to focus and hence further accelerate the calculation. Furthemore, several groups have developed multi-term interaction potentials and others use multi-copy approaches to simulate protein flexibility, which both add to the computational cost of FFT-based docking algorithms. Hence there is a need to develop more powerful and more versatile FFT docking techniques.
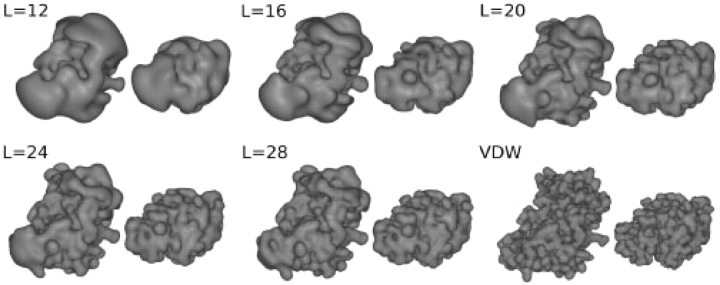
Results: This article presents a closed-form 6D spherical polar Fourier correlation expression from which arbitrary multi-dimensional multi-property multi-resolution FFT correlations may be generated. The approach is demonstrated by calculating 1D, 3D and 5D rotational correlations of 3D shape and electrostatic expansions up to polynomial order L=30 on a 2 GB personal computer. As expected, 3D correlations are found to be considerably faster than 1D correlations but, surprisingly, 5D correlations are often slower than 3D correlations. Nonetheless, we show that 5D correlations will be advantageous when calculating multi-term knowledge-based interaction potentials. When docking the 84 complexes of the Protein Docking Benchmark, blind 3D shape plus electrostatic correlations take around 30 minutes on a contemporary personal computer and find acceptable solutions within the top 20 in 16 cases. Applying a simple angular constraint to focus the calculation around the receptor binding site produces acceptable solutions within the top 20 in 28 cases. Further constraining the search to the ligand binding site gives up to 48 solutions within the top 20, with calculation times of just a few minutes per complex. Hence the approach described provides a practical and fast tool for rigid body protein-protein docking, especially when prior knowledge about one or both binding sites is available.
Figures


Similar articles
-
Protein docking using spherical polar Fourier correlations.Proteins. 2000 May 1;39(2):178-94. Proteins. 2000. PMID: 10737939
-
On the computation of molecular surface correlations for protein docking using fourier techniques.J Bioinform Comput Biol. 2007 Aug;5(4):915-35. doi: 10.1142/s0219720007002916. J Bioinform Comput Biol. 2007. PMID: 17787063
-
Protein-protein docking by fast generalized Fourier transforms on 5D rotational manifolds.Proc Natl Acad Sci U S A. 2016 Jul 26;113(30):E4286-93. doi: 10.1073/pnas.1603929113. Epub 2016 Jul 13. Proc Natl Acad Sci U S A. 2016. PMID: 27412858 Free PMC article.
-
Rigid-Docking Approaches to Explore Protein-Protein Interaction Space.Adv Biochem Eng Biotechnol. 2017;160:33-55. doi: 10.1007/10_2016_41. Adv Biochem Eng Biotechnol. 2017. PMID: 27830312 Review.
-
Predicting 3D structures of protein-protein complexes.Curr Pharm Biotechnol. 2008 Apr;9(2):57-66. doi: 10.2174/138920108783955209. Curr Pharm Biotechnol. 2008. PMID: 18393862 Review.
Cited by
-
Solution structures, dynamics, and ice growth inhibitory activity of peptide fragments derived from an antarctic yeast protein.PLoS One. 2012;7(11):e49788. doi: 10.1371/journal.pone.0049788. Epub 2012 Nov 28. PLoS One. 2012. PMID: 23209600 Free PMC article.
-
Structural basis for ligand recognition and activation of RAGE.Structure. 2010 Oct 13;18(10):1342-52. doi: 10.1016/j.str.2010.05.017. Structure. 2010. PMID: 20947022 Free PMC article.
-
"Redirecting an anti-IL-1β antibody to bind a new, unrelated and computationally predicted epitope on hIL-17A".Commun Biol. 2023 Sep 29;6(1):997. doi: 10.1038/s42003-023-05369-x. Commun Biol. 2023. PMID: 37773269 Free PMC article.
-
Structures of sequential open states in a symmetrical opening transition of the TolC exit duct.Proc Natl Acad Sci U S A. 2011 Feb 1;108(5):2112-7. doi: 10.1073/pnas.1012588108. Epub 2011 Jan 18. Proc Natl Acad Sci U S A. 2011. PMID: 21245342 Free PMC article.
-
In Silico Insights into HIV-1 Vpu-Tetherin Interactions and Its Mutational Counterparts.Med Sci (Basel). 2019 Jun 22;7(6):74. doi: 10.3390/medsci7060074. Med Sci (Basel). 2019. PMID: 31234536 Free PMC article.
References
-
- Berman HM, et al. The protein data bank. Acta. Cryst. 2002;D58:899–907. - PubMed
-
- Biedenharn LC, Louck J.C.1. Angular Momentum in Quantum Physics. Reading, MA: Addison-Wesley; 1981.
-
- Chen R, et al. ZDOCK: an initial-stage protein-docking algorithm. Proteins Struct. Funct. Genet. 2003;52:80–87. - PubMed
-
- Dominguez C, et al. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003;125:1731–1737. - PubMed
-
- Edmonds AR. Angular Momentum in Quantum Physics. New Jersey: Princeton University Press; 1957.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
