

Review
doi: 10.1146/annurev.genom.9.081307.164303.
Disorders of lysosome-related organelle biogenesis: clinical and molecular genetics
Affiliations
- PMID: 18544035
- PMCID: PMC2755194
- DOI: 10.1146/annurev.genom.9.081307.164303
Item in Clipboard
Review
Disorders of lysosome-related organelle biogenesis: clinical and molecular genetics
Annu Rev Genomics Hum Genet.
2008.
Abstract
Lysosome-related organelles (LROs) are a heterogeneous group of vesicles that share various features with lysosomes, but are distinct in function, morphology, and composition. The biogenesis of LROs employs a common machinery, and genetic defects in this machinery can affect all LROs or only an individual LRO, resulting in a variety of clinical features. In this review, we discuss the main components of LRO biogenesis. We also summarize the function, composition, and resident cell types of the major LROs. Finally, we describe the clinical characteristics of the major human LRO disorders.
Figures
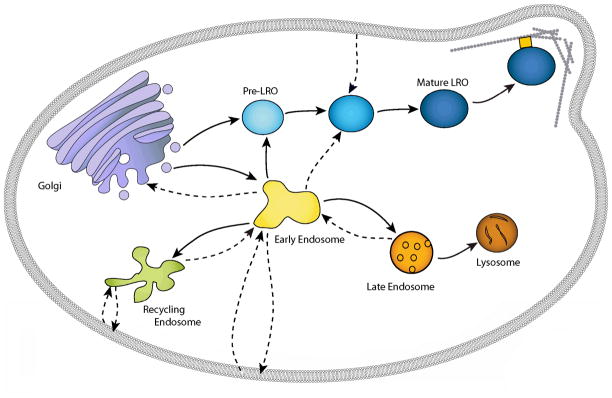
The endosomal system is a collection of highly dynamic compartments defined by their morphology, contingent of marker proteins, function and accessibility to endocytic tracers. Rather than focus on a specific cell type, this model shows the basic endosomal elements involved in generic LRO biogenesis. Solid arrows depict maturation/genesis of a compartment while dashed arrows represent the movement of cargo. The early (sorting) endosome is the major sorting center of the cell and is defined by the presence of EEA1 and Rab5 and by rapid accessibility to endocytic tracers (5–15 min). Sorting of Golgi derived biosynthetic cargo destined for LROs or late endosomes/lysosomes as well as receptors and other molecules internalized from the cell membrane occurs in the early endosome. Some receptors return to the cell membrane by way of recycling endosomes, while others continue on to late endosomes and lysosomes for degradation. As the early endosome matures, intralumenal vesicles containing proteins targeted for lysosomal degradation accumulate and the multivesicular body (MVB)/late endosome is formed. The MVB/late endosome is the last site of sorting and is defined by its morphology and the presence of MPRs and LBPA. MPRs deliver lysosomal hydrolases and are then recycled from the MVB/late endosome back to the TGN. MVBs/late endosomes fuse with (or mature into) lysosomes to deliver their contents for degradation. Lysosomes lack both intralumenal vesicles and MPRs and are considered end-stage degradative compartments. Specialized LROs co-exist in the same cell as non-specialized lysosomes and contents destined for each compartment must be sorted appropriately. In some LRO containing cells, LRO formation involves sequential delivery of LRO-specific proteins. A subset of proteins destined for LROs (e.g. PMEL17 in melanocytes) are sorted from a post-Golgi compartment to an endosomal intermediate and drive the formation of a pre-LRO (e.g. stage I/II melanosome). A second set of LRO specific proteins (e.g. TYR and TYRP1) are sorted from the early endosome to the pre-LRO compartment, resulting in a mature LRO. The mature LRO then acquires specific accessory proteins (Rabs, motor proteins, SNAREs) that assist in its function and/or localization.
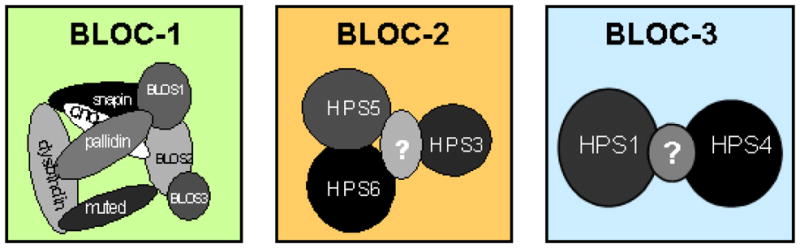
The currently recognized proteins identified as components of each BLOC are indicated. Most of the BLOC subunits are associated with subtypes of Hermansky-Pudlak syndrome. Subunits indicated by “?” are predicted by molecular weight analyses of the entire complex, and may represent a yet-to-be-identified subunit.
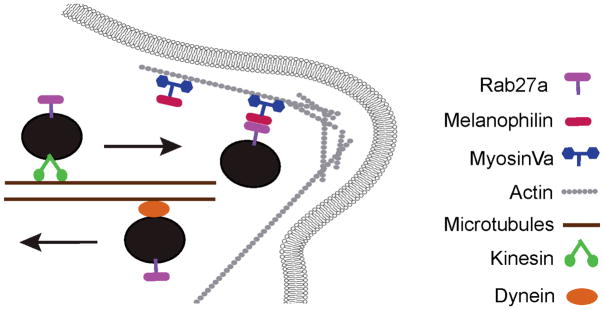
In melanocytes, Rab27a resides on mature melanosomes which travel to the cell periphery on microtubules via a kinesin motor protein. Once in the periphery, Rab27a recruits the motor protein myosinVa via a direct interaction with Melanophilin. The Rab27a-Melanophilin-MyosinVa tripartite complex is then responsible for the accumulation of mature melanosomes in the actin-rich dendritic tips. This localization is necessary for efficient transfer of melanosomes to keratinocytes for normal pigmentation. If any member of the tripartite complex is defective, melanosomes are not captured in the periphery and return to the cell center on microtubules in a dynein-mediated process. Defects in any member of the tripartite complex result in Griscelli syndrome.
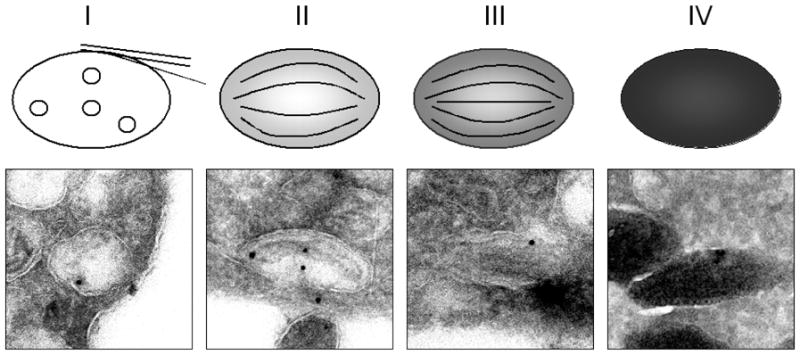
The upper panel represents a cartoon of the four stages of melanosome biogenesis. The lower panel shows an immune-electron microscopy study of human primary epidermal melanocytes. Ultrathin cryosections labeled with anti-NKI beteb, a PMEL17-specific marker, conjugated to 20-nm gold particles. Stage I melanosomes, electron-lucent MVBs without melanin, contain multiple intralumenal vesicles. Stage II melanosomes are more elongated with PMEL17-containing intralumenal striations that run the length of the organelle. Stage III melanosomes exhibit melanin deposits on the striations. These striations are masked in stage IV melanosomes, due to complete melanization.
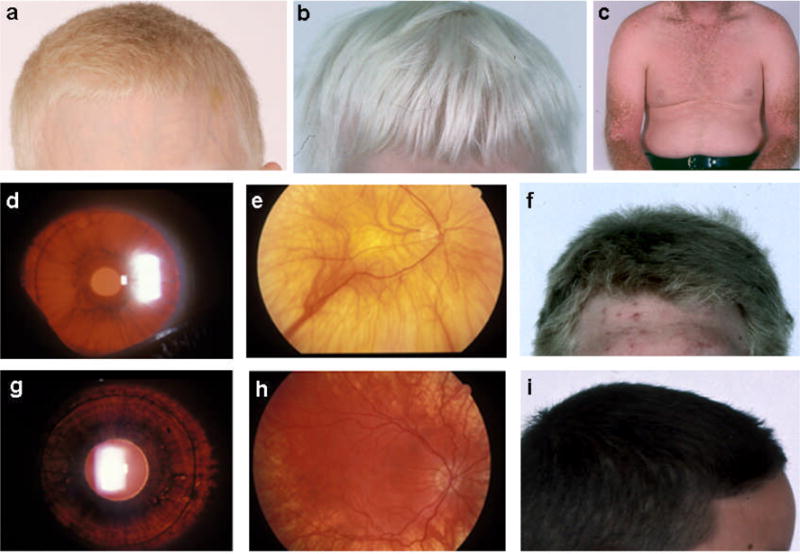
(a) Light colored hair of a boy with HPS-1. (b) White hair of a boy with HPS-4. (c) Keratoses in sun-exposed areas of a Puerto Rican patient with HPS-1. (d) Significant iris transillumination in an HPS-1 patient. Orange light, abnormally present, appears because the iris contains insufficient melanin to block it. (e) Pale retinal fundus in HPS-1, with vessels clearly visible due to lack of retinal pigment epithelium. (f) Tan-blond hair of a patient with HPS-2. (g) Mild iris transillumination in a patient with HPS-3. (h) Mild hypopigmentation of the retina in HPS-3. (i) Dark hair of a Puerto Rican boy with HPS-3. [Figures d, e, g, and h courtesy of Dr. E. Tsilou, National Eye Institute].
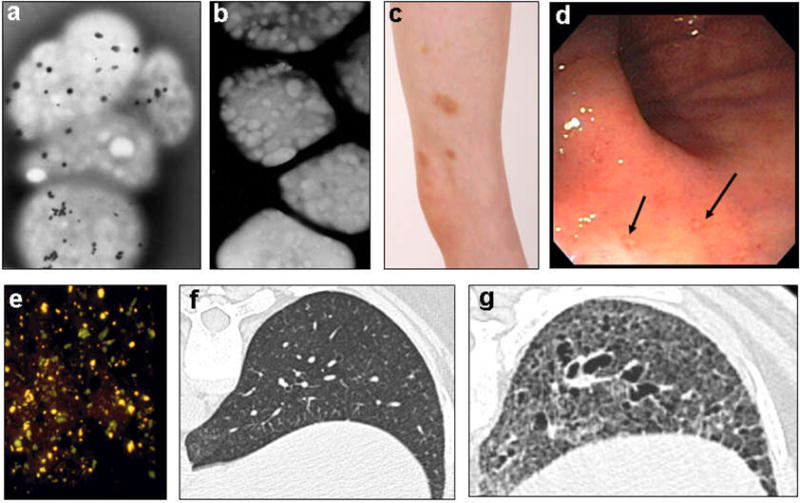
(a) Whole mount electron micrographs of normal platelets, each contain several delta granules (small black dots). (b) HPS platelets, devoid of delta granules. (c) Spontaneous bruising in a boy with HPS. (d) HPS intestine showing mucosal ulcerations (arrows). (e) Autofluorescent ceroid lipofucsin (yellow) in renal tubular cells of an HPS-1 patient sloughed into the urine. (f) High-resolution CT scan of a normal lung. (g) High-resolution CT scan of an HPS-1 lung, showing bullae, fibrosis, and loss of alveoli. [(a) and (b) courtesy of Dr. James G. White, University of Minnesota; (f) and (g) courtesy of Dr. Thomas Markello, NHGRI].
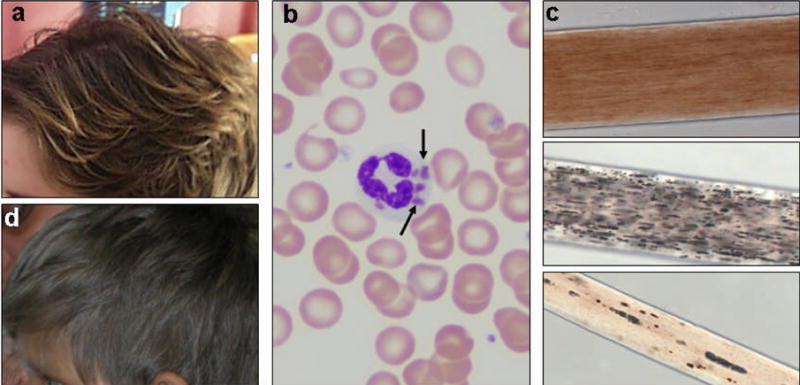
(a) Light brown hair in a girl with CHS, after bone marrow transplantation. (b) Wright stain of a peripheral blood smear from a classic CHS patient, showing abnormal giant granules in a polymorphonuclear leucocyte (arrows). (c) Light microscopy of hair shafts from a normal individual (top), a patient with CHS (middle), and a patient with GS2 (bottom). Uneven granularity characterizes the pigmentation pattern of the bottom two hair shafts. (d) Silvery gray hair of a boy with GS2.
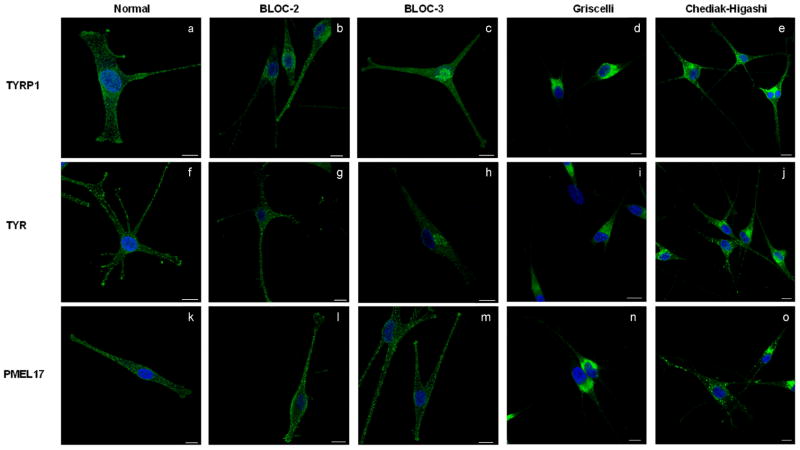
Confocal immunofluorescence microscopy of primary epidermal melanocytes derived from a normally pigmented individual and patients with LRO biogenesis defects stained for (a–e) TYRP1 (anti-mouse MEL5), (f–j) TYR (anti-mouse Tyrosinase), and (k–o) PMEL17 (anti-mouse HMB45). All melanocytes were co-stained with TO-PRO-3 to visualize the nucleus. (a,f,k) Normal melanocytes demonstrate punctuate (a) TYRP1, (f) TYR and (k) PMEL17 staining in the perinuclear region and throughout the dendrites with TYRP1 and TYR accumulation in the dendritic tips. (b,g,l) BLOC-2 deficient melanocytes were derived from an HPS-5 patient compound heterozygous for a nonsense mutation (c.2624C>T; p.R865X) and a one base pair deletion (c.2264delT: P.L875fsX19) in exon 18 of HPS5. BLOC-2 deficient melanocytes display punctate staining of (b) TYRP1 and (g) TYR in the perinuclear region extending into the dendrites, but lack pronounced accumulation of TYRP1 and TYR in the tips. (l) PMEL17 staining in BLOC-2 deficient melanocytes is distributed throughout the cell as in normal melanocytes. (c,h,m) BLOC-3 deficient melanocytes were derived from an HPS-1 patient homozygous for a 16 base-pair duplication (c.1472_1487dup16) in exon 15 of HPS1. In the absence of BLOC-3 (c) TYRP1 and (h) TYR are concentrated in the perinuclear/TGN region while (m) PMEL17 extends into the dendrites and appears normally distributed. (d,i,n) GS2 melanocytes were derived from a patient compound heterozygous for two nonsense mutations (c.550C>T; p.R184X and c.598C>T; p.R200X) in Rab27A. In GS2 melanocytes, (d) TYRP1, (i) TYR, and (n) PMEL17 localize to the perinuclear area and do not occupy the dendritic tips. (e,j,o) CHS melanocytes were derived from a patient carrying a nonsense mutation (c.1540C>T; p.R514X) in exon 5 and a one base pair deletion (c.9893delT; p.F3298fsX3304) in exon 43 in CHS1. In CHS melanocytes, (e) TYRP1 localizes to vesicular structures in the perinuclear area with some dendritic localization, (j) TYR accumulates in large granules that localize to the dendrites but not their tips, and (o) PMEL17 localizes to enlarged vesicular structures that are present in the perinuclear and dendritic area but not in the dendritic tips. All images are 1D projections of confocal z-sections. Scale bars = 10 μm.
Similar articles
-
Lysosome-related organelles.FASEB J. 2000 Jul;14(10):1265-78. doi: 10.1096/fj.14.10.1265. FASEB J. 2000. PMID: 10877819 Review.
-
Insights into the biogenesis of lysosome-related organelles from the study of the Hermansky-Pudlak syndrome.Ann N Y Acad Sci. 2004 Dec;1038:103-14. doi: 10.1196/annals.1315.018. Ann N Y Acad Sci. 2004. PMID: 15838104
-
Genetic regulation of Caenorhabditis elegans lysosome related organelle function.PLoS Genet. 2013 Oct;9(10):e1003908. doi: 10.1371/journal.pgen.1003908. Epub 2013 Oct 24. PLoS Genet. 2013. PMID: 24204312 Free PMC article.
-
Hermansky-Pudlak syndrome and Chediak-Higashi syndrome: disorders of vesicle formation and trafficking.Thromb Haemost. 2001 Jul;86(1):233-45. Thromb Haemost. 2001. PMID: 11487012 Review.
-
Hermansky-Pudlak syndrome: pigmentary and non-pigmentary defects and their pathogenesis.Pigment Cell Melanoma Res. 2013 Mar;26(2):176-92. doi: 10.1111/pcmr.12051. Epub 2012 Dec 31. Pigment Cell Melanoma Res. 2013. PMID: 23171219 Review.
Cited by
-
Mouse RC/BTB2, a member of the RCC1 superfamily, localizes to spermatid acrosomal vesicles.PLoS One. 2012;7(6):e39846. doi: 10.1371/journal.pone.0039846. Epub 2012 Jun 29. PLoS One. 2012. PMID: 22768142 Free PMC article.
-
The WASH complex, an endosomal Arp2/3 activator, interacts with the Hermansky-Pudlak syndrome complex BLOC-1 and its cargo phosphatidylinositol-4-kinase type IIα.Mol Biol Cell. 2013 Jul;24(14):2269-84. doi: 10.1091/mbc.E13-02-0088. Epub 2013 May 15. Mol Biol Cell. 2013. PMID: 23676666 Free PMC article.
-
Acrosome biogenesis: Revisiting old questions to yield new insights.Spermatogenesis. 2011 Apr;1(2):95-98. doi: 10.4161/spmg.1.2.16820. Spermatogenesis. 2011. PMID: 22319656 Free PMC article.
-
Drosophila mauve mutants reveal a role of LYST homologs late in the maturation of phagosomes and autophagosomes.Traffic. 2012 Dec;13(12):1680-92. doi: 10.1111/tra.12005. Epub 2012 Sep 20. Traffic. 2012. PMID: 22934826 Free PMC article.
-
Assembly of the biogenesis of lysosome-related organelles complex-3 (BLOC-3) and its interaction with Rab9.J Biol Chem. 2010 Mar 5;285(10):7794-804. doi: 10.1074/jbc.M109.069088. Epub 2010 Jan 4. J Biol Chem. 2010. PMID: 20048159 Free PMC article.
References
-
- Ali B, Seabra M. Targeting of Rab GTPases to cellular membranes. Biochem Soc Trans. 2005;33:652–6. - PubMed
-
- Anderson PD, Huizing M, Claassen DA, White J, Gahl WA. Hermansky-Pudlak syndrome type 4 (HPS-4): clinical and molecular characteristics. Hum Genet. 2003;113:10–7. - PubMed
-
- Anikster Y, Huizing M, White J, Shevchenko YO, Fitzpatrick DL, et al. Mutation of a new gene causes a unique form of Hermansky-Pudlak syndrome in a genetic isolate of central Puerto Rico. Nat Genet. 2001;28:376–80. - PubMed
-
- Barral DC, Seabra MC. The melanosome as a model to study organelle motility in mammals. Pigment Cell Res. 2004;17:111–8. - PubMed
-
- Berson JF, Theos AC, Harper DC, Tenza D, Raposo G, Marks MS. Proprotein convertase cleavage liberates a fibrillogenic fragment of a resident glycoprotein to initiate melanosome biogenesis. J Cell Biol. 2003;161:521–33. This manuscript describes MVBs as intermediates in the generation of stage II melanosomes, and it shows that PMEL17 is the main component of the intralumenal fibrils of stage II melanosomes. These fibrils are generated by cleavage of PMEL17 in a post-Golgi compartment. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
