

Membrane traffic and turnover in TRP-ML1-deficient cells: a revised model for mucolipidosis type IV pathogenesis
- PMID: 18504305
- PMCID: PMC2413042
- DOI: 10.1084/jem.20072194
Membrane traffic and turnover in TRP-ML1-deficient cells: a revised model for mucolipidosis type IV pathogenesis
Abstract
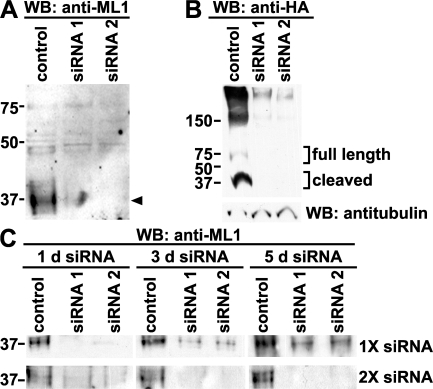
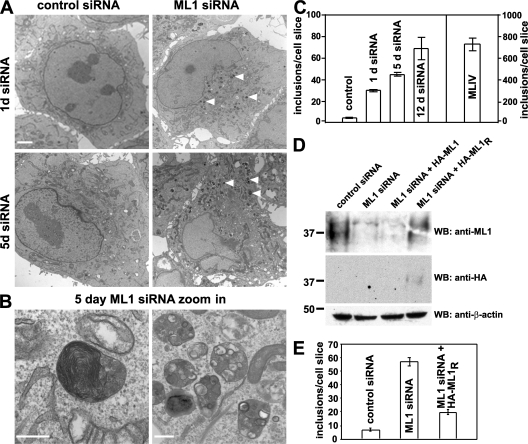
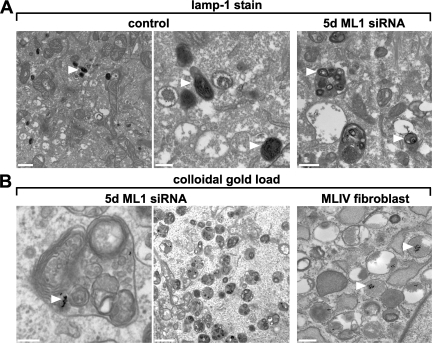
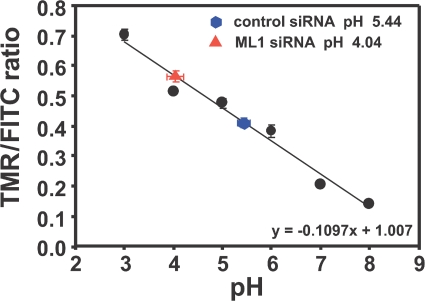
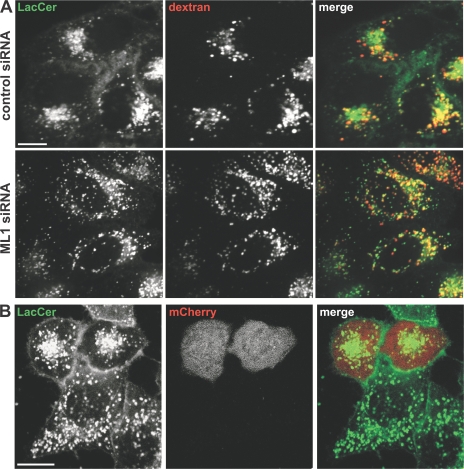
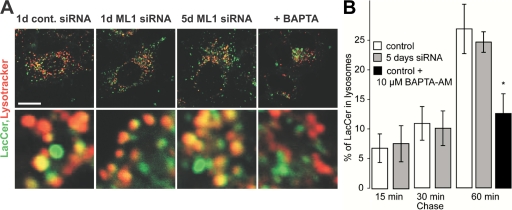
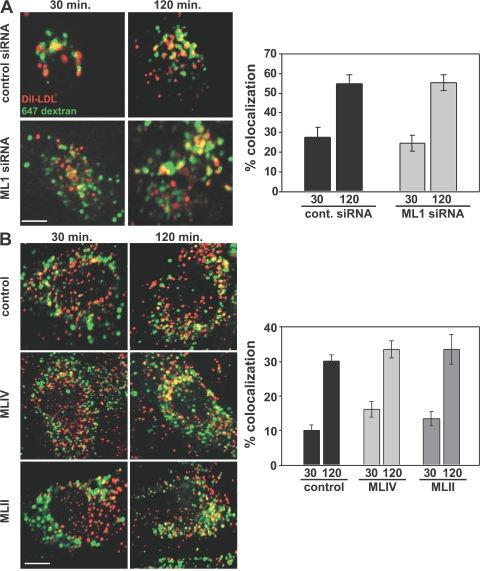
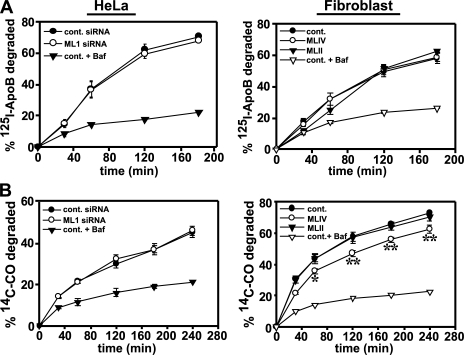
The lysosomal storage disorder mucolipidosis type IV (MLIV) is caused by mutations in the transient receptor potential-mucolipin-1 (TRP-ML1) ion channel. The "biogenesis" model for MLIV pathogenesis suggests that TRP-ML1 modulates postendocytic delivery to lysosomes by regulating interactions between late endosomes and lysosomes. This model is based on observed lipid trafficking delays in MLIV patient fibroblasts. Because membrane traffic aberrations may be secondary to lipid buildup in chronically TRP-ML1-deficient cells, we depleted TRP-ML1 in HeLa cells using small interfering RNA and examined the effects on cell morphology and postendocytic traffic. TRP-ML1 knockdown induced gradual accumulation of membranous inclusions and, thus, represents a good model in which to examine the direct effects of acute TRP-ML1 deficiency on membrane traffic. Ratiometric imaging revealed decreased lysosomal pH in TRP-ML1-deficient cells, suggesting a disruption in lysosomal function. Nevertheless, we found no effect of TRP-ML1 knockdown on the kinetics of protein or lipid delivery to lysosomes. In contrast, by comparing degradation kinetics of low density lipoprotein constituents, we confirmed a selective defect in cholesterol but not apolipoprotein B hydrolysis in MLIV fibroblasts. We hypothesize that the effects of TRP-ML1 loss on hydrolytic activity have a cumulative effect on lysosome function, resulting in a lag between TRP-ML1 loss and full manifestation of MLIV.
Figures
Similar articles
-
TRP-ML1 regulates lysosomal pH and acidic lysosomal lipid hydrolytic activity.J Biol Chem. 2006 Mar 17;281(11):7294-301. doi: 10.1074/jbc.M508211200. Epub 2005 Dec 16. J Biol Chem. 2006. PMID: 16361256
-
Reconstitution of lysosomal NAADP-TRP-ML1 signaling pathway and its function in TRP-ML1(-/-) cells.Am J Physiol Cell Physiol. 2011 Aug;301(2):C421-30. doi: 10.1152/ajpcell.00393.2010. Epub 2011 May 25. Am J Physiol Cell Physiol. 2011. PMID: 21613607 Free PMC article.
-
Molecular pathophysiology of mucolipidosis type IV: pH dysregulation of the mucolipin-1 cation channel.Hum Mol Genet. 2004 Mar 15;13(6):617-27. doi: 10.1093/hmg/ddh067. Epub 2004 Jan 28. Hum Mol Genet. 2004. PMID: 14749347
-
Cation channel activity of mucolipin-1: the effect of calcium.Pflugers Arch. 2005 Oct;451(1):304-12. doi: 10.1007/s00424-005-1448-9. Epub 2005 Aug 23. Pflugers Arch. 2005. PMID: 16133264 Review.
-
Mucolipin 1: endocytosis and cation channel--a review.Pflugers Arch. 2005 Oct;451(1):313-7. doi: 10.1007/s00424-004-1361-7. Epub 2004 Nov 27. Pflugers Arch. 2005. PMID: 15570434 Review.
Cited by
-
Identification of the penta-EF-hand protein ALG-2 as a Ca2+-dependent interactor of mucolipin-1.J Biol Chem. 2009 Dec 25;284(52):36357-36366. doi: 10.1074/jbc.M109.047241. Epub 2009 Oct 28. J Biol Chem. 2009. PMID: 19864416 Free PMC article.
-
Mucolipins: Intracellular TRPML1-3 channels.FEBS Lett. 2010 May 17;584(10):2013-21. doi: 10.1016/j.febslet.2009.12.056. Epub 2010 Jan 13. FEBS Lett. 2010. PMID: 20074572 Free PMC article. Review.
-
TRP channels of intracellular membranes.J Neurochem. 2010 Apr;113(2):313-28. doi: 10.1111/j.1471-4159.2010.06626.x. Epub 2010 Jan 28. J Neurochem. 2010. PMID: 20132470 Free PMC article. Review.
-
Autophagy inhibition mediated by MCOLN1/TRPML1 suppresses cancer metastasis via regulating a ROS-driven TP53/p53 pathway.Autophagy. 2022 Aug;18(8):1932-1954. doi: 10.1080/15548627.2021.2008752. Epub 2021 Dec 8. Autophagy. 2022. PMID: 34878954 Free PMC article.
-
Biphasic regulation of lysosomal exocytosis by oxidative stress.Cell Calcium. 2016 Nov;60(5):356-362. doi: 10.1016/j.ceca.2016.08.002. Epub 2016 Aug 29. Cell Calcium. 2016. PMID: 27593159 Free PMC article.
References
-
- Bargal, R., N. Avidan, E. Ben-Asher, Z. Olender, M. Zeigler, A. Frumkin, A. Raas-Rothschild, G. Glusman, D. Lancet, and G. Bach. 2000. Identification of the gene causing mucolipidosis type IV. Nat. Genet. 26:118–123. - PubMed
-
- Sun, M., E. Goldin, S. Stahl, J.L. Falardeau, J.C. Kennedy, J.S. Acierno Jr., C. Bove, C.R. Kaneski, J. Nagle, M.C. Bromley, et al. 2000. Mucolipidosis type IV is caused by mutations in a gene encoding a novel transient receptor potential channel. Hum. Mol. Genet. 9:2471–2478. - PubMed
-
- Bach, G. 2001. Mucolipidosis type IV. Mol. Genet. Metab. 73:197–203. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
