

The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice
- PMID: 18497889
- PMCID: PMC2391284
- DOI: 10.1172/JCI33585
The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice
Abstract
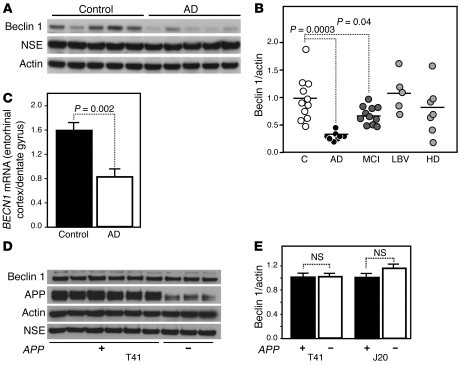
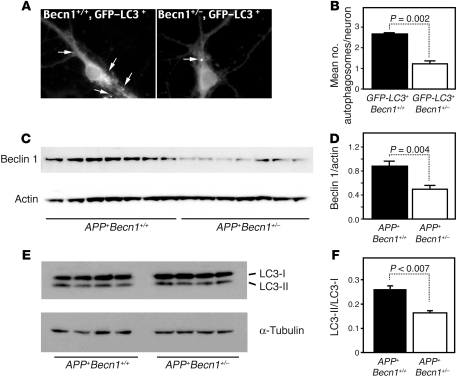
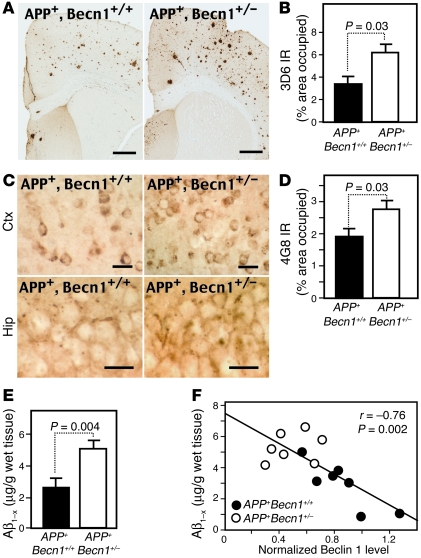
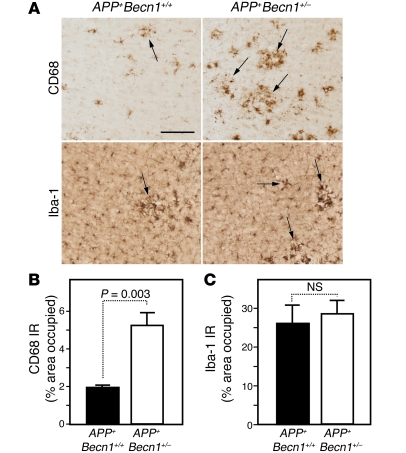
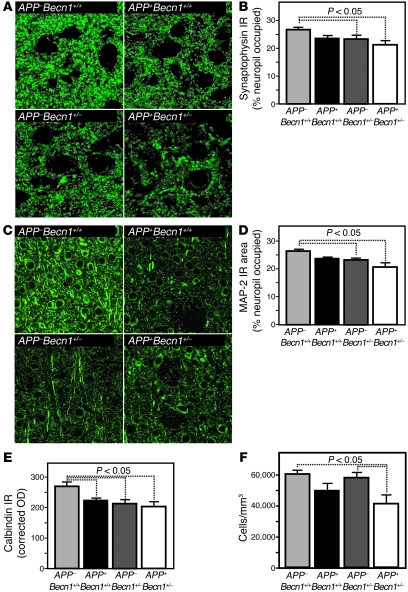
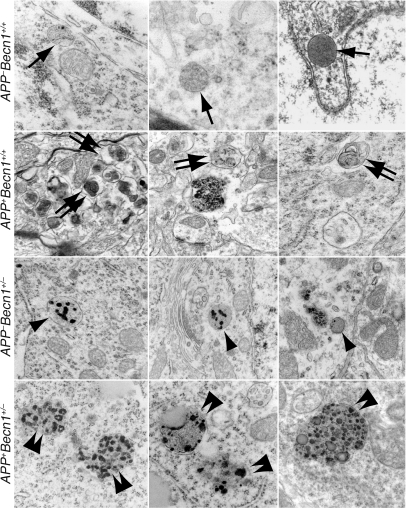
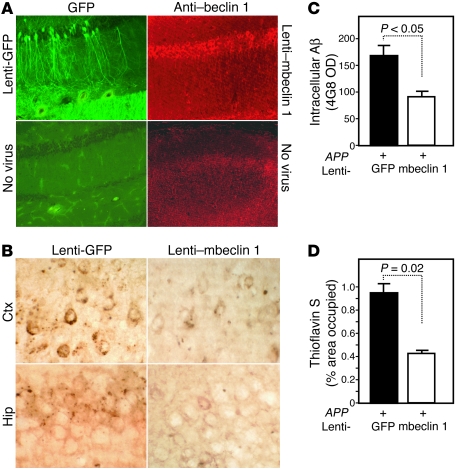
Autophagy is the principal cellular pathway for degradation of long-lived proteins and organelles and regulates cell fate in response to stress. Recently, autophagy has been implicated in neurodegeneration, but whether it is detrimental or protective remains unclear. Here we report that beclin 1, a protein with a key role in autophagy, was decreased in affected brain regions of patients with Alzheimer disease (AD) early in the disease process. Heterozygous deletion of beclin 1 (Becn1) in mice decreased neuronal autophagy and resulted in neurodegeneration and disruption of lysosomes. In transgenic mice that express human amyloid precursor protein (APP), a model for AD, genetic reduction of Becn1 expression increased intraneuronal amyloid beta (Abeta) accumulation, extracellular Abeta deposition, and neurodegeneration and caused microglial changes and profound neuronal ultrastructural abnormalities. Administration of a lentiviral vector expressing beclin 1 reduced both intracellular and extracellular amyloid pathology in APP transgenic mice. We conclude that beclin 1 deficiency disrupts neuronal autophagy, modulates APP metabolism, and promotes neurodegeneration in mice and that increasing beclin 1 levels may have therapeutic potential in AD.
Figures
Comment in
-
Regulation of Abeta pathology by beclin 1: a protective role for autophagy?J Clin Invest. 2008 Jun;118(6):2015-8. doi: 10.1172/JCI35662. J Clin Invest. 2008. PMID: 18497881 Free PMC article.
Similar articles
-
Regulation of Abeta pathology by beclin 1: a protective role for autophagy?J Clin Invest. 2008 Jun;118(6):2015-8. doi: 10.1172/JCI35662. J Clin Invest. 2008. PMID: 18497881 Free PMC article.
-
Impaired autophagy and APP processing in Alzheimer's disease: The potential role of Beclin 1 interactome.Prog Neurobiol. 2013 Jul-Aug;106-107:33-54. doi: 10.1016/j.pneurobio.2013.06.002. Epub 2013 Jul 1. Prog Neurobiol. 2013. PMID: 23827971 Review.
-
Regulation of amyloid precursor protein processing by the Beclin 1 complex.PLoS One. 2010 Jun 15;5(6):e11102. doi: 10.1371/journal.pone.0011102. PLoS One. 2010. PMID: 20559548 Free PMC article.
-
Mesenchymal stem cells enhance autophagy and increase β-amyloid clearance in Alzheimer disease models.Autophagy. 2014 Jan;10(1):32-44. doi: 10.4161/auto.26508. Epub 2013 Jan 1. Autophagy. 2014. PMID: 24149893 Free PMC article.
-
Effects of CX3CR1 and Fractalkine Chemokines in Amyloid Beta Clearance and p-Tau Accumulation in Alzheimer's Disease (AD) Rodent Models: Is Fractalkine a Systemic Biomarker for AD?Curr Alzheimer Res. 2016;13(4):403-12. doi: 10.2174/1567205013666151116125714. Curr Alzheimer Res. 2016. PMID: 26567742 Review.
Cited by
-
Transcription Factor EB Is Selectively Reduced in the Nuclear Fractions of Alzheimer's and Amyotrophic Lateral Sclerosis Brains.Neurosci J. 2016;2016:4732837. doi: 10.1155/2016/4732837. Epub 2016 Jun 28. Neurosci J. 2016. PMID: 27433468 Free PMC article.
-
Protein homeostasis, aging and Alzheimer's disease.Mol Neurobiol. 2012 Aug;46(1):41-54. doi: 10.1007/s12035-012-8246-0. Epub 2012 Feb 24. Mol Neurobiol. 2012. PMID: 22361852 Free PMC article. Review.
-
Intersection between Redox Homeostasis and Autophagy: Valuable Insights into Neurodegeneration.Antioxidants (Basel). 2021 Apr 28;10(5):694. doi: 10.3390/antiox10050694. Antioxidants (Basel). 2021. PMID: 33924878 Free PMC article. Review.
-
Treatment with Trehalose Prevents Behavioral and Neurochemical Deficits Produced in an AAV α-Synuclein Rat Model of Parkinson's Disease.Mol Neurobiol. 2016 May;53(4):2258-68. doi: 10.1007/s12035-015-9173-7. Epub 2015 May 14. Mol Neurobiol. 2016. PMID: 25972237
-
Loss of endophilin-B1 exacerbates Alzheimer's disease pathology.Brain. 2015 Jul;138(Pt 7):2005-19. doi: 10.1093/brain/awv128. Epub 2015 May 16. Brain. 2015. PMID: 25981964 Free PMC article.
References
-
- Levine B. Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell. 2005;120:159–162. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- AG10435/AG/NIA NIH HHS/United States
- R01 CA084254/CA/NCI NIH HHS/United States
- R01 AG020603/AG/NIA NIH HHS/United States
- R01 AG018440/AG/NIA NIH HHS/United States
- CA84254/CA/NCI NIH HHS/United States
- AG20603/AG/NIA NIH HHS/United States
- AG02270/AG/NIA NIH HHS/United States
- AG22074/AG/NIA NIH HHS/United States
- AG5131/AG/NIA NIH HHS/United States
- P50 AG005131/AG/NIA NIH HHS/United States
- R01 AG030144/AG/NIA NIH HHS/United States
- R37 AG018440/AG/NIA NIH HHS/United States
- P01 AG010435/AG/NIA NIH HHS/United States
- AG18440/AG/NIA NIH HHS/United States
- P01 AG022074/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
