

3' adenylation determines mRNA abundance and monitors completion of RNA editing in T. brucei mitochondria
- PMID: 18464794
- PMCID: PMC2426725
- DOI: 10.1038/emboj.2008.87
3' adenylation determines mRNA abundance and monitors completion of RNA editing in T. brucei mitochondria
Abstract
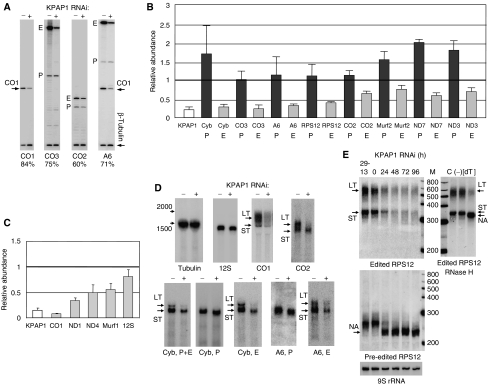
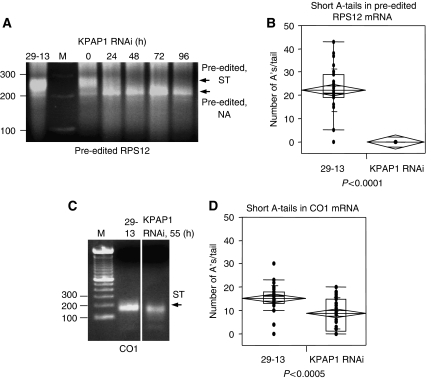
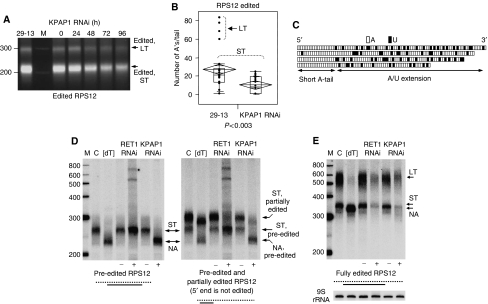
Expression of the mitochondrial genome in protozoan parasite Trypanosoma brucei is controlled post-transcriptionally and requires extensive U-insertion/deletion mRNA editing. In mitochondrial extracts, 3' adenylation reportedly influences degradation kinetics of synthetic edited and pre-edited mRNAs. We have identified and characterized a mitochondrial poly(A) polymerase, termed KPAP1, and determined major polypeptides in the polyadenylation complex. Inhibition of KPAP1 expression abrogates short and long A-tails typically found in mitochondrial mRNAs, and decreases the abundance of never-edited and edited transcripts. Pre-edited mRNAs are not destabilized by the lack of 3' adenylation, whereas short A-tails are required and sufficient to maintain the steady-state levels of partially edited, fully edited, and never-edited mRNAs. The editing directed by a single guide RNA is sufficient to impose a requirement for the short A-tail in edited molecules. Upon completion of the editing process, the short A-tails are extended as (A/U) heteropolymers into structures previously thought to be long poly(A) tails. These data provide the first direct evidence of functional interactions between 3' processing and editing of mitochondrial mRNAs in trypanosomes.
Figures
Similar articles
-
Poly(A) binding KPAF4/5 complex stabilizes kinetoplast mRNAs in Trypanosoma brucei.Nucleic Acids Res. 2020 Sep 4;48(15):8645-8662. doi: 10.1093/nar/gkaa575. Nucleic Acids Res. 2020. PMID: 32614436 Free PMC article.
-
Targeted depletion of a mitochondrial nucleotidyltransferase suggests the presence of multiple enzymes that polymerize mRNA 3' tails in Trypanosoma brucei mitochondria.Mol Biochem Parasitol. 2007 Aug;154(2):158-69. doi: 10.1016/j.molbiopara.2007.04.014. Epub 2007 Apr 27. Mol Biochem Parasitol. 2007. PMID: 17543398 Free PMC article.
-
Pentatricopeptide repeat proteins stimulate mRNA adenylation/uridylation to activate mitochondrial translation in trypanosomes.Mol Cell. 2011 Apr 8;42(1):106-17. doi: 10.1016/j.molcel.2011.02.021. Mol Cell. 2011. PMID: 21474072 Free PMC article.
-
Lexis and Grammar of Mitochondrial RNA Processing in Trypanosomes.Trends Parasitol. 2020 Apr;36(4):337-355. doi: 10.1016/j.pt.2020.01.006. Epub 2020 Feb 28. Trends Parasitol. 2020. PMID: 32191849 Free PMC article. Review.
-
U-Insertion/Deletion mRNA-Editing Holoenzyme: Definition in Sight.Trends Parasitol. 2016 Feb;32(2):144-156. doi: 10.1016/j.pt.2015.10.004. Epub 2015 Nov 10. Trends Parasitol. 2016. PMID: 26572691 Free PMC article. Review.
Cited by
-
REH2 RNA helicase in kinetoplastid mitochondria: ribonucleoprotein complexes and essential motifs for unwinding and guide RNA (gRNA) binding.J Biol Chem. 2010 Jan 8;285(2):1220-8. doi: 10.1074/jbc.M109.051862. Epub 2009 Oct 22. J Biol Chem. 2010. PMID: 19850921 Free PMC article.
-
Kinetoplastid guide RNA biogenesis is dependent on subunits of the mitochondrial RNA binding complex 1 and mitochondrial RNA polymerase.RNA. 2009 Apr;15(4):588-99. doi: 10.1261/rna.1411809. Epub 2009 Feb 18. RNA. 2009. PMID: 19228586 Free PMC article.
-
Uridine insertion/deletion RNA editing in trypanosomatid mitochondria: In search of the editosome.RNA. 2009 Jul;15(7):1338-44. doi: 10.1261/rna.1642809. Epub 2009 May 15. RNA. 2009. PMID: 19447916 Free PMC article.
-
PPR polyadenylation factor defines mitochondrial mRNA identity and stability in trypanosomes.EMBO J. 2017 Aug 15;36(16):2435-2454. doi: 10.15252/embj.201796808. Epub 2017 Jul 6. EMBO J. 2017. PMID: 28684539 Free PMC article.
-
Comparison of the Mitochondrial Genomes and Steady State Transcriptomes of Two Strains of the Trypanosomatid Parasite, Leishmania tarentolae.PLoS Negl Trop Dis. 2015 Jul 23;9(7):e0003841. doi: 10.1371/journal.pntd.0003841. eCollection 2015. PLoS Negl Trop Dis. 2015. PMID: 26204118 Free PMC article.
References
-
- Abraham J, Feagin J, Stuart K (1988) Characterization of cytochrome c oxidase III transcripts that are edited only in the 3′ region. Cell 55: 267–272 - PubMed
-
- Aphasizhev R, Aphasizheva I (2007) RNA editing uridylyltransferases of trypanosomatids. Methods Enzymol 424: 51–67 - PubMed
-
- Aphasizhev R, Aphasizheva I, Simpson L (2004) Multiple terminal uridylyltransferases of trypanosomes. FEBS Lett 572: 15–18 - PubMed
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
