

Hyperglycemia-induced reactive oxygen species toxicity to endothelial cells is dependent on paracrine mediators
- PMID: 18420487
- PMCID: PMC2453610
- DOI: 10.2337/db07-1520
Hyperglycemia-induced reactive oxygen species toxicity to endothelial cells is dependent on paracrine mediators
Abstract
Objective: This study determined the effects of high glucose exposure and cytokine treatment on generation of reactive oxygen species (ROS) and activation of inflammatory and apoptotic pathways in human retinal endothelial cells (HRECs).
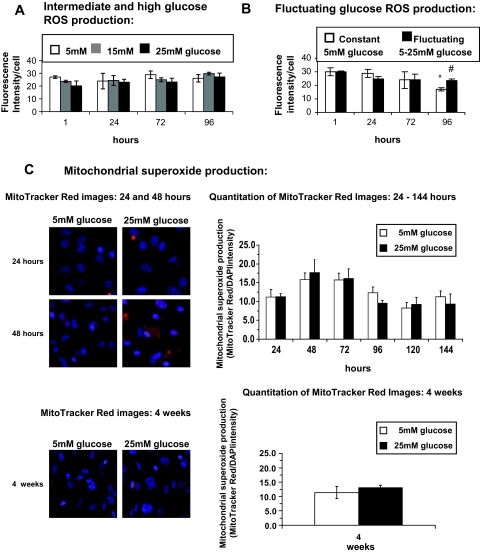
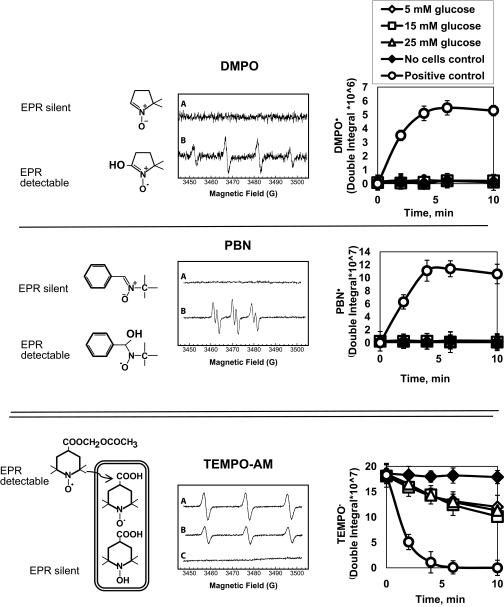
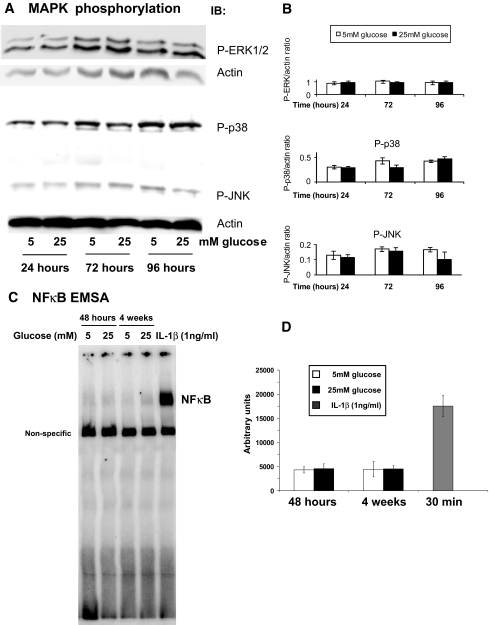
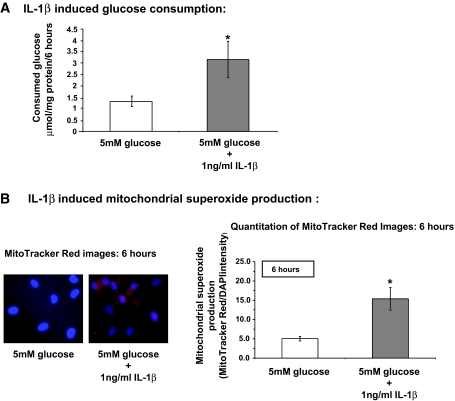
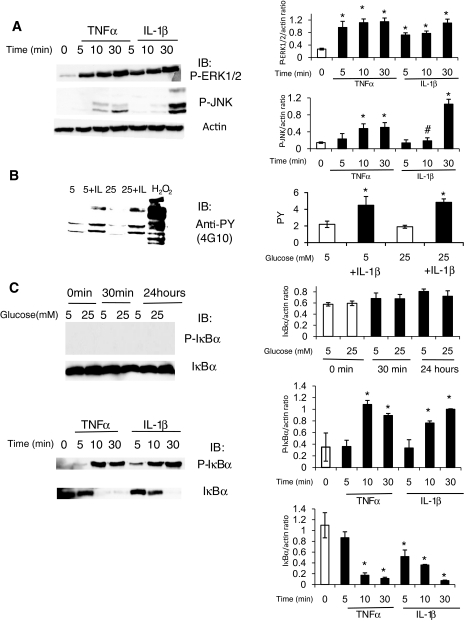
Research design and methods: Glucose consumption of HRECs, human retinal pigment epithelial cells (HRPEs), and human Müller cells (HMCs) under elevated glucose conditions was measured and compared with cytokine treatment. Production of ROS in HRECs was examined using 5-(and-6)-chloromethyl-2',7'-dichlorodihydrofluorescein diacetate (CM-H(2)DCFDA), spin-trap electron paramagnetic resonance, and MitoTracker Red staining after high glucose and cytokine treatment. The activation of different signaling cascades, including the mitogen-activated protein kinase pathways, tyrosine phosphorylation pathways, and apoptosis by high glucose and cytokines in HRECs, was determined.
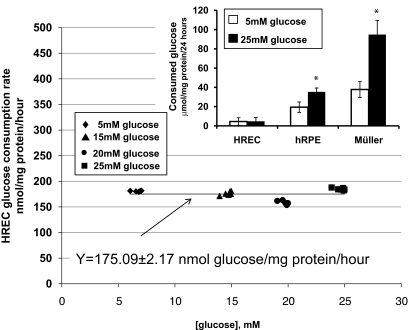
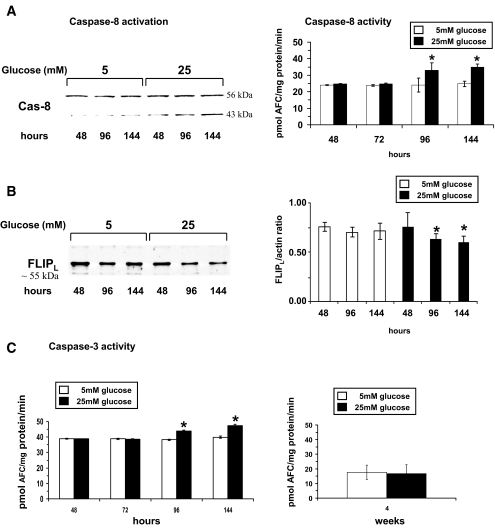
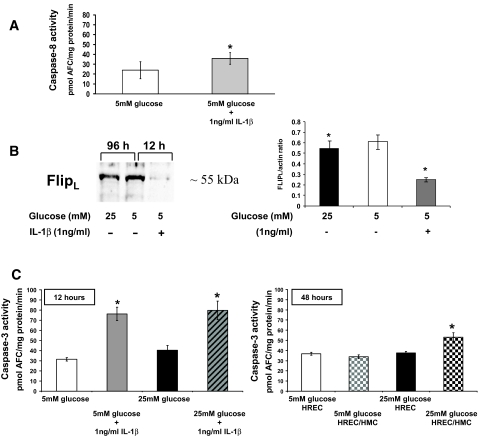
Results: HRECs, in contrast to HRPEs and HMCs, did not increase glucose consumption in response to increasing glucose concentrations. Exposure of HRECs to 25 mmol/l glucose did not stimulate endogenous ROS production, activation of nuclear factor-kappaB (NF-kappaB), extracellular signal-related kinase (ERK), p38 and Jun NH(2)-terminal kinase (JNK), tyrosine phosphorylation, interleukin (IL)-1beta, or tumor necrosis factor-alpha (TNF-alpha) production and only slightly affected apoptotic cell death pathways compared with normal glucose (5 mmol/l). In marked contrast, exposure of HRECs to proinflammatory cytokines IL-1beta or TNF-alpha increased glucose consumption, mitochondrial superoxide production, ERK and JNK phosphorylation, tyrosine phosphorylation, NF-kappaB activation, and caspase activation.
Conclusions: Our in vitro results indicate that HRECs respond to cytokines rather than high glucose, suggesting that in vivo diabetes-related endothelial injury in the retina may be due to glucose-induced cytokine release by other retinal cells and not a direct effect of high glucose.
Figures
Similar articles
-
Bradykinin alleviates DR retinal endothelial injury by regulating HMGB-1/NF-κB pathway.Eur Rev Med Pharmacol Sci. 2019 Jul;23(13):5535-5541. doi: 10.26355/eurrev_201907_18286. Eur Rev Med Pharmacol Sci. 2019. PMID: 31298304
-
Natural flavonoid galangin alleviates microglia-trigged blood-retinal barrier dysfunction during the development of diabetic retinopathy.J Nutr Biochem. 2019 Mar;65:1-14. doi: 10.1016/j.jnutbio.2018.11.006. Epub 2018 Dec 4. J Nutr Biochem. 2019. PMID: 30597356
-
Intermittent high glucose enhances cell proliferation and VEGF expression in retinal endothelial cells: the role of mitochondrial reactive oxygen species.Mol Cell Biochem. 2010 Oct;343(1-2):27-35. doi: 10.1007/s11010-010-0495-5. Epub 2010 Jun 4. Mol Cell Biochem. 2010. PMID: 20524146
-
The effect of reactive oxygen species on the synthesis of prostanoids from arachidonic acid.J Physiol Pharmacol. 2013 Aug;64(4):409-21. J Physiol Pharmacol. 2013. PMID: 24101387 Review.
-
Hypoxia and oxidative stress in the causation of diabetic retinopathy.Curr Diabetes Rev. 2011 Sep;7(5):291-304. doi: 10.2174/157339911797415620. Curr Diabetes Rev. 2011. PMID: 21916837 Review.
Cited by
-
Endothelial cell and platelet bioenergetics: effect of glucose and nutrient composition.PLoS One. 2012;7(6):e39430. doi: 10.1371/journal.pone.0039430. Epub 2012 Jun 22. PLoS One. 2012. PMID: 22745753 Free PMC article.
-
The pathogenesis of early retinal changes of diabetic retinopathy.Doc Ophthalmol. 2012 Feb;124(1):15-26. doi: 10.1007/s10633-011-9305-y. Doc Ophthalmol. 2012. PMID: 22302291 Review.
-
Interleukin-1β and mitochondria damage, and the development of diabetic retinopathy.J Ocul Biol Dis Infor. 2011 Jun;4(1-2):3-9. doi: 10.1007/s12177-011-9074-6. Epub 2011 Dec 28. J Ocul Biol Dis Infor. 2011. PMID: 23272269 Free PMC article.
-
Mitochondrial Oxidative Stress, Mitochondrial DNA Damage and Their Role in Age-Related Vascular Dysfunction.Int J Mol Sci. 2015 Jul 13;16(7):15918-53. doi: 10.3390/ijms160715918. Int J Mol Sci. 2015. PMID: 26184181 Free PMC article. Review.
-
The importance of mitochondria in age-related and inherited eye disorders.Ophthalmic Res. 2010;44(3):179-90. doi: 10.1159/000316480. Epub 2010 Sep 9. Ophthalmic Res. 2010. PMID: 20829642 Free PMC article. Review.
References
-
- Baynes JW: Role of oxidative stress in development of complications in diabetes. Diabetes 40 :405 –412,1991 - PubMed
-
- Koya D, King GL: Protein kinase C activation and the development of diabetic complications. Diabetes 47 :859 –866,1998 - PubMed
-
- Vinores SA, Campochiaro PA, Williams EH, May EE, Green WR, Sorenson RL: Aldose reductase expression in human diabetic retina and retinal pigment epithelium. Diabetes 37 :1658 –1664,1988 - PubMed
-
- Brownlee M: Lilly Lecture 1993: Glycation and diabetic complications. Diabetes 43 :836 –841,1994 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
