

A map of human protein interactions derived from co-expression of human mRNAs and their orthologs
- PMID: 18414481
- PMCID: PMC2387231
- DOI: 10.1038/msb.2008.19
A map of human protein interactions derived from co-expression of human mRNAs and their orthologs
Abstract
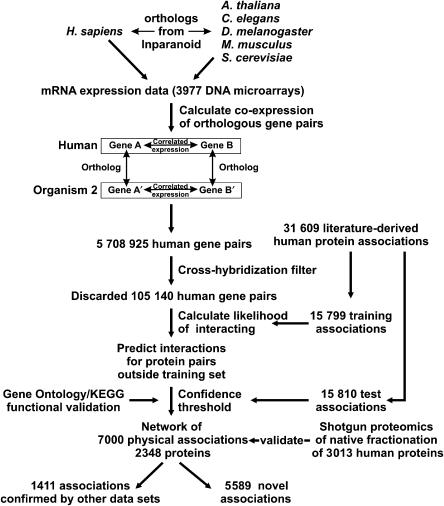
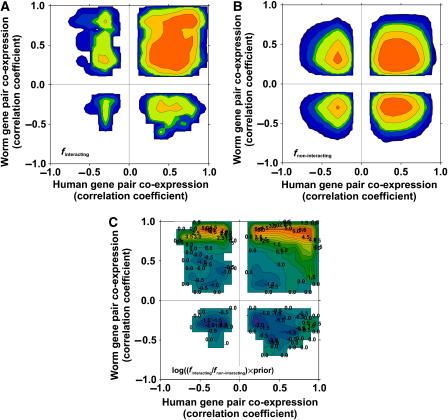
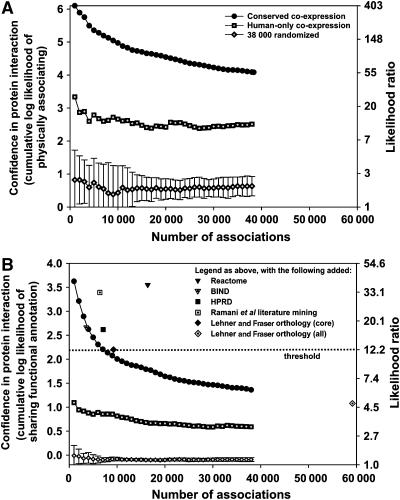
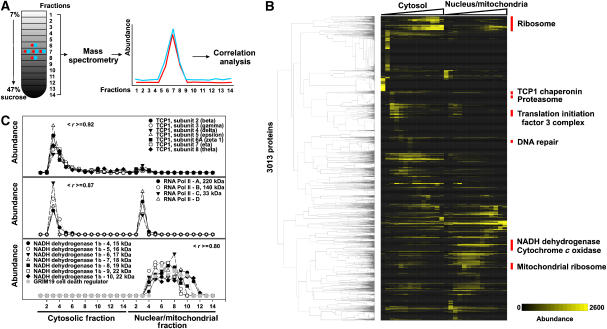
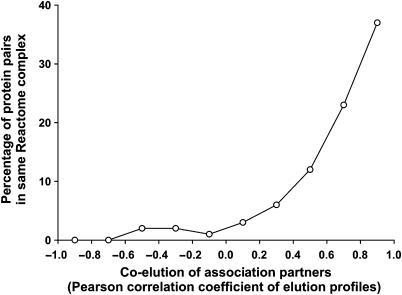
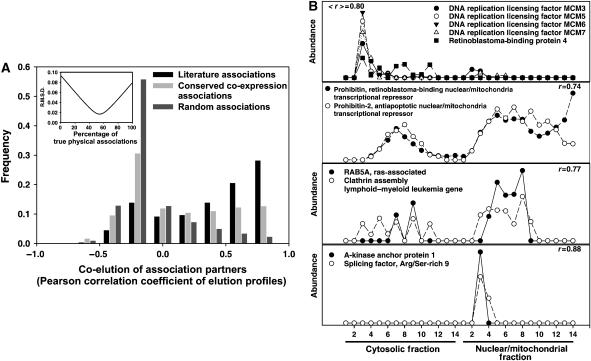
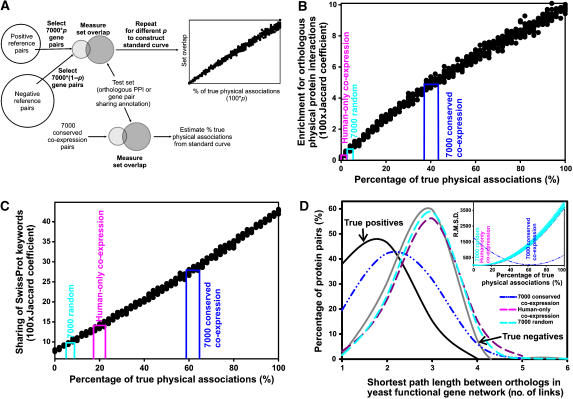
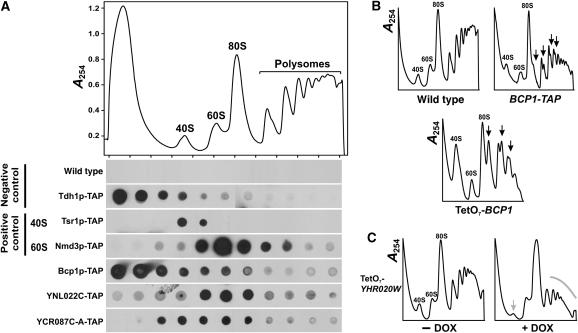
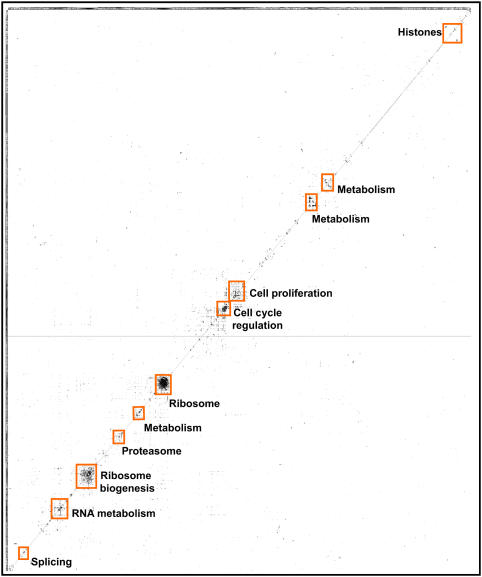
The human protein interaction network will offer global insights into the molecular organization of cells and provide a framework for modeling human disease, but the network's large scale demands new approaches. We report a set of 7000 physical associations among human proteins inferred from indirect evidence: the comparison of human mRNA co-expression patterns with those of orthologous genes in five other eukaryotes, which we demonstrate identifies proteins in the same physical complexes. To evaluate the accuracy of the predicted physical associations, we apply quantitative mass spectrometry shotgun proteomics to measure elution profiles of 3013 human proteins during native biochemical fractionation, demonstrating systematically that putative interaction partners tend to co-sediment. We further validate uncharacterized proteins implicated by the associations in ribosome biogenesis, including WBSCR20C, associated with Williams-Beuren syndrome. This meta-analysis therefore exploits non-protein-based data, but successfully predicts associations, including 5589 novel human physical protein associations, with measured accuracies of 54+/-10%, comparable to direct large-scale interaction assays. The new associations' derivation from conserved in vivo phenomena argues strongly for their biological relevance.
Figures
Similar articles
-
A multi-model statistical approach for proteomic spectral count quantitation.J Proteomics. 2016 Jul 20;144:23-32. doi: 10.1016/j.jprot.2016.05.032. Epub 2016 May 31. J Proteomics. 2016. PMID: 27260494 Free PMC article.
-
Novel approaches to map protein interactions.Curr Opin Biotechnol. 2003 Feb;14(1):119-25. doi: 10.1016/s0958-1669(02)00005-8. Curr Opin Biotechnol. 2003. PMID: 12566011 Review.
-
Mass spectrometry and the search for moonlighting proteins.Mass Spectrom Rev. 2005 Nov-Dec;24(6):772-82. doi: 10.1002/mas.20041. Mass Spectrom Rev. 2005. PMID: 15605385 Review.
-
Comparative proteomic and transcriptomic profiling of the fission yeast Schizosaccharomyces pombe.Mol Syst Biol. 2007;3:79. doi: 10.1038/msb4100117. Epub 2007 Feb 13. Mol Syst Biol. 2007. PMID: 17299416 Free PMC article.
-
Probabilistic model of the human protein-protein interaction network.Nat Biotechnol. 2005 Aug;23(8):951-9. doi: 10.1038/nbt1103. Nat Biotechnol. 2005. PMID: 16082366
Cited by
-
Methylation of ribosomal RNA by NSUN5 is a conserved mechanism modulating organismal lifespan.Nat Commun. 2015 Jan 30;6:6158. doi: 10.1038/ncomms7158. Nat Commun. 2015. PMID: 25635753 Free PMC article.
-
ORTom: a multi-species approach based on conserved co-expression to identify putative functional relationships among genes in tomato.Plant Mol Biol. 2010 Jul;73(4-5):519-32. doi: 10.1007/s11103-010-9638-z. Epub 2010 Apr 22. Plant Mol Biol. 2010. PMID: 20411302
-
A complex network approach reveals a pivotal substructure of genes linked to schizophrenia.PLoS One. 2018 Jan 5;13(1):e0190110. doi: 10.1371/journal.pone.0190110. eCollection 2018. PLoS One. 2018. PMID: 29304112 Free PMC article.
-
LDGIdb: a database of gene interactions inferred from long-range strong linkage disequilibrium between pairs of SNPs.BMC Res Notes. 2012 May 2;5:212. doi: 10.1186/1756-0500-5-212. BMC Res Notes. 2012. PMID: 22551073 Free PMC article.
-
Functional genome annotation by combined analysis across microarray studies of Trypanosoma brucei.PLoS Negl Trop Dis. 2010 Aug 31;4(8):e810. doi: 10.1371/journal.pntd.0000810. PLoS Negl Trop Dis. 2010. PMID: 20824174 Free PMC article.
References
-
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25: 25–29 - PMC - PubMed
-
- Ball CA, Awad IA, Demeter J, Gollub J, Hebert JM, Hernandez-Boussard T, Jin H, Matese JC, Nitzberg M, Wymore F, Zachariah ZK, Brown PO, Sherlock G (2005) The Stanford Microarray Database accommodates additional microarray platforms and data formats. Nucleic Acids Res 33: (Database issue) D580–D582 - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
