

Opposing effects of polyglutamine expansion on native protein complexes contribute to SCA1
- PMID: 18337722
- PMCID: PMC2377396
- DOI: 10.1038/nature06731
Opposing effects of polyglutamine expansion on native protein complexes contribute to SCA1
Abstract
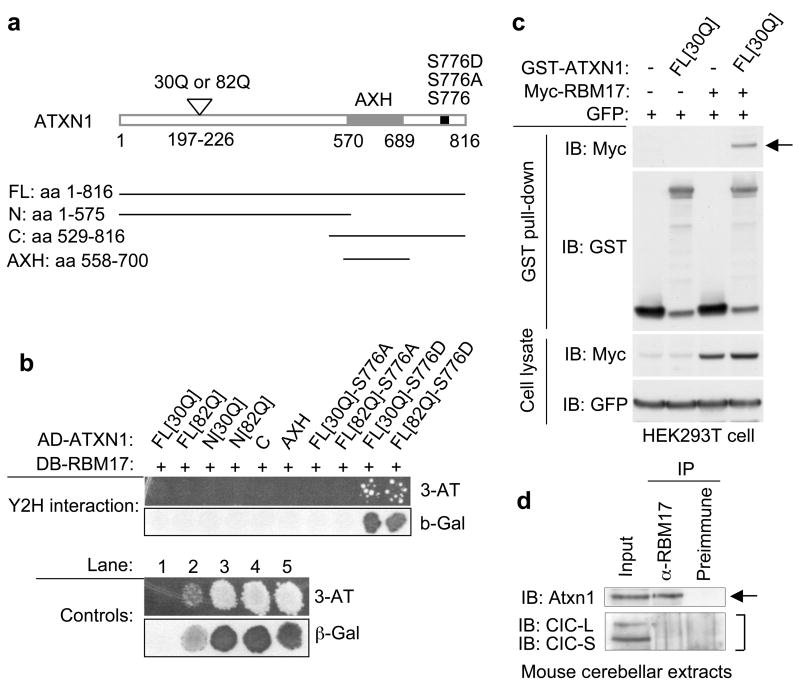
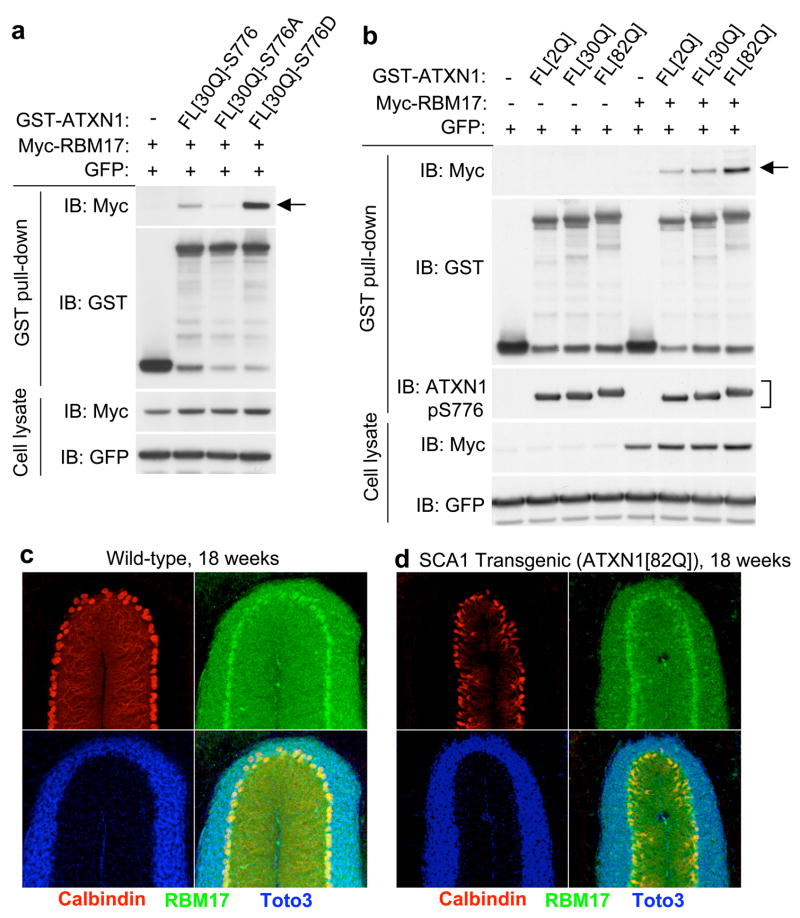
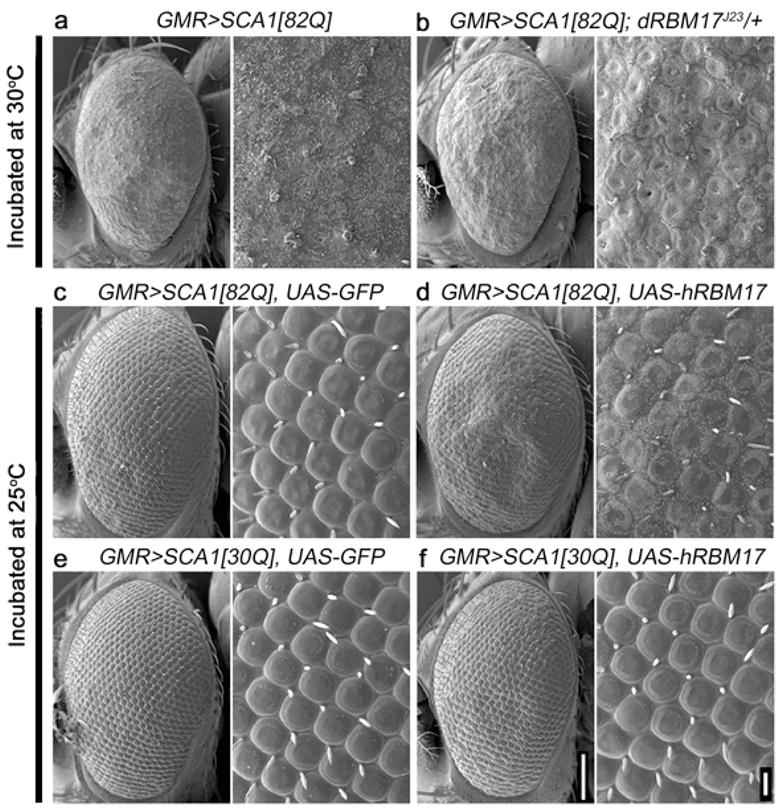
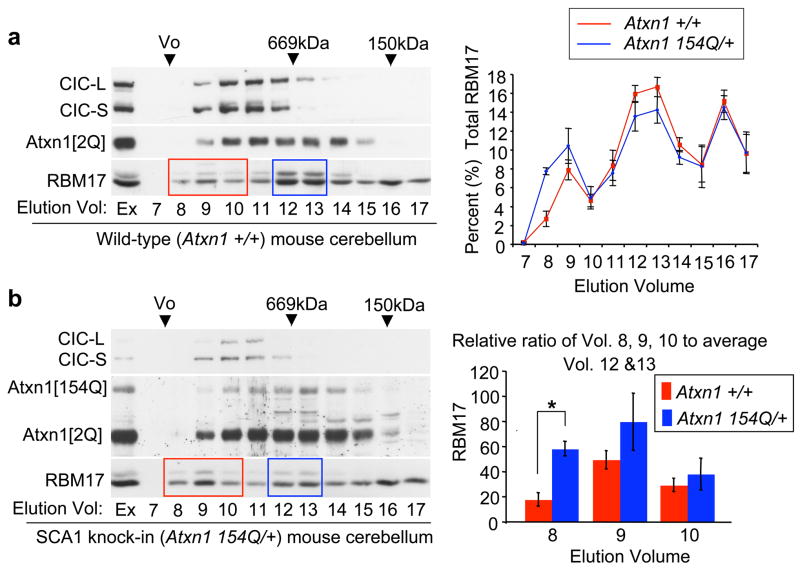
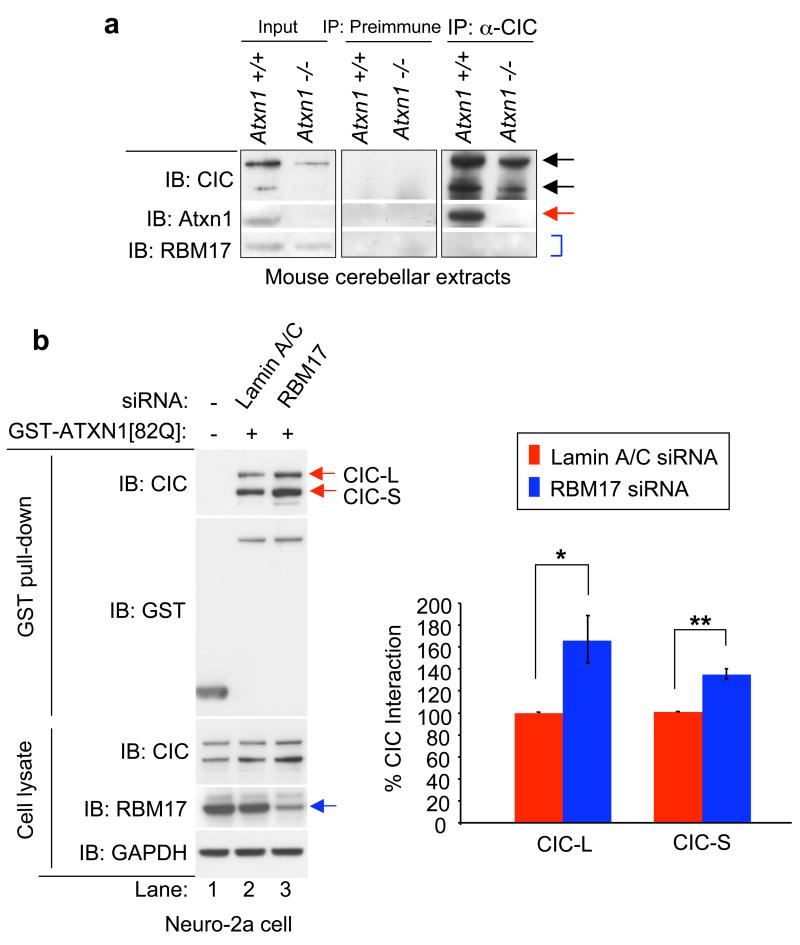
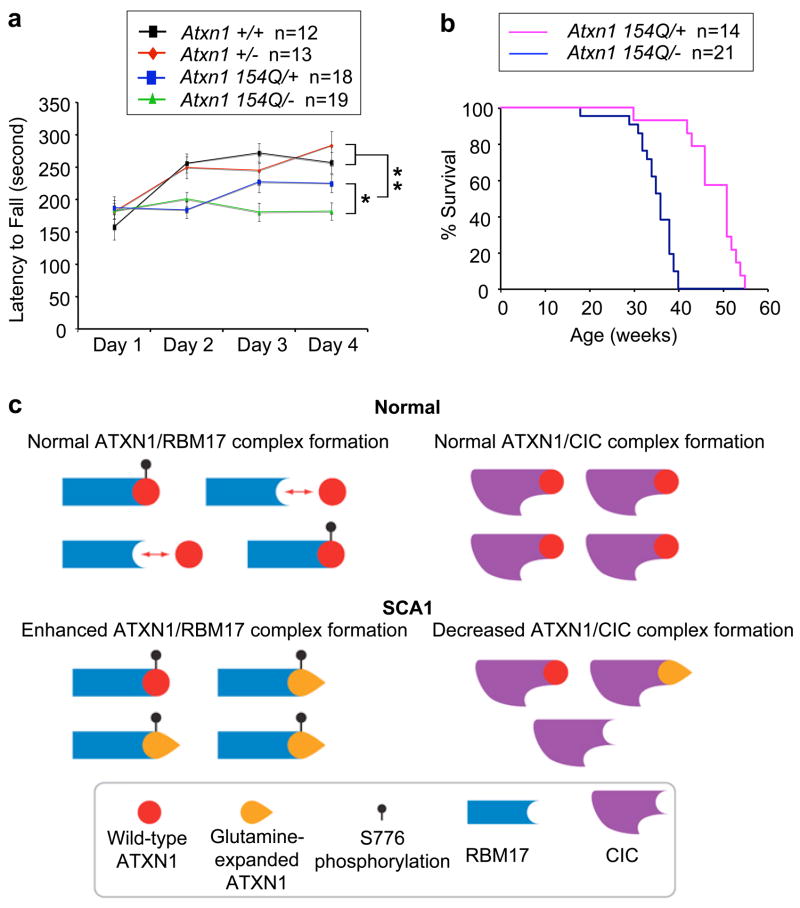
Spinocerebellar ataxia type 1 (SCA1) is a dominantly inherited neurodegenerative disease caused by expansion of a glutamine-encoding repeat in ataxin 1 (ATXN1). In all known polyglutamine diseases, the glutamine expansion confers toxic functions onto the protein; however, the mechanism by which this occurs remains enigmatic, in light of the fact that the mutant protein apparently maintains interactions with its usual partners. Here we show that the expanded polyglutamine tract differentially affects the function of the host protein in the context of different endogenous protein complexes. Polyglutamine expansion in ATXN1 favours the formation of a particular protein complex containing RBM17, contributing to SCA1 neuropathology by means of a gain-of-function mechanism. Concomitantly, polyglutamine expansion attenuates the formation and function of another protein complex containing ATXN1 and capicua, contributing to SCA1 through a partial loss-of-function mechanism. This model provides mechanistic insight into the molecular pathogenesis of SCA1 as well as other polyglutamine diseases.
Figures
Comment in
-
Neurodegeneration: a question of balance.Nature. 2008 Apr 10;452(7188):707-8. doi: 10.1038/452707a. Nature. 2008. PMID: 18401401 No abstract available.
Similar articles
-
Neurodegeneration: a question of balance.Nature. 2008 Apr 10;452(7188):707-8. doi: 10.1038/452707a. Nature. 2008. PMID: 18401401 No abstract available.
-
Pathogenic mechanisms of a polyglutamine-mediated neurodegenerative disease, spinocerebellar ataxia type 1.J Biol Chem. 2009 Mar 20;284(12):7425-9. doi: 10.1074/jbc.R800041200. Epub 2008 Oct 28. J Biol Chem. 2009. PMID: 18957430 Free PMC article. Review.
-
Structural basis of the phosphorylation dependent complex formation of neurodegenerative disease protein Ataxin-1 and RBM17.Biochem Biophys Res Commun. 2014 Jul 11;449(4):399-404. doi: 10.1016/j.bbrc.2014.05.063. Epub 2014 May 22. Biochem Biophys Res Commun. 2014. PMID: 24858692
-
Duplication of Atxn1l suppresses SCA1 neuropathology by decreasing incorporation of polyglutamine-expanded ataxin-1 into native complexes.Nat Genet. 2007 Mar;39(3):373-9. doi: 10.1038/ng1977. Epub 2007 Feb 18. Nat Genet. 2007. PMID: 17322884
-
SCA1-phosphorylation, a regulator of Ataxin-1 function and pathogenesis.Prog Neurobiol. 2012 Dec;99(3):179-85. doi: 10.1016/j.pneurobio.2012.04.003. Epub 2012 Apr 16. Prog Neurobiol. 2012. PMID: 22531670 Free PMC article. Review.
Cited by
-
PRMT5- mediated symmetric arginine dimethylation is attenuated by mutant huntingtin and is impaired in Huntington's disease (HD).Cell Cycle. 2015;14(11):1716-29. doi: 10.1080/15384101.2015.1033595. Cell Cycle. 2015. PMID: 25927346 Free PMC article.
-
Gene, Stem Cell, and Alternative Therapies for SCA 1.Front Mol Neurosci. 2016 Aug 12;9:67. doi: 10.3389/fnmol.2016.00067. eCollection 2016. Front Mol Neurosci. 2016. PMID: 27570504 Free PMC article. Review.
-
An autism-linked missense mutation in SHANK3 reveals the modularity of Shank3 function.Mol Psychiatry. 2020 Oct;25(10):2534-2555. doi: 10.1038/s41380-018-0324-x. Epub 2019 Jan 4. Mol Psychiatry. 2020. PMID: 30610205 Free PMC article.
-
ATXN1 N-terminal region explains the binding differences of wild-type and expanded forms.BMC Med Genomics. 2019 Oct 26;12(1):145. doi: 10.1186/s12920-019-0594-4. BMC Med Genomics. 2019. PMID: 31655597 Free PMC article.
-
Complementary proteomics strategies capture an ataxin-1 interactome in Neuro-2a cells.Sci Data. 2018 Nov 20;5:180262. doi: 10.1038/sdata.2018.262. Sci Data. 2018. PMID: 30457570 Free PMC article.
References
-
- Orr HT, Zoghbi HY. Trinucleotide Repeat Disorders. Annu Rev Neurosci. 2007;30:575–621. - PubMed
-
- Harjes P, Wanker EE. The hunt for huntingtin function: interaction partners tell many different stories. Trends Biochem Sci. 2003;28:425–33. - PubMed
-
- Li SH, Li XJ. Huntingtin-protein interactions and the pathogenesis of Huntington’s disease. Trends Genet. 2004;20:146–54. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
