

Bioinformatic and functional analysis of RNA secondary structure elements among different genera of human and animal caliciviruses
- PMID: 18319285
- PMCID: PMC2377429
- DOI: 10.1093/nar/gkn096
Bioinformatic and functional analysis of RNA secondary structure elements among different genera of human and animal caliciviruses
Abstract
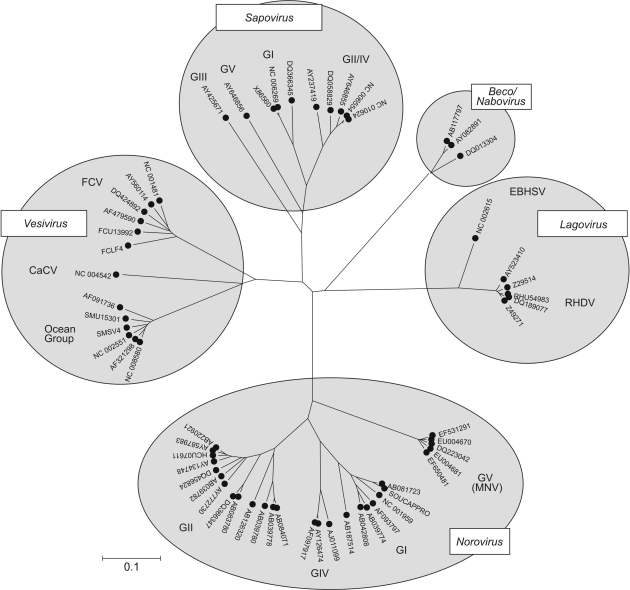
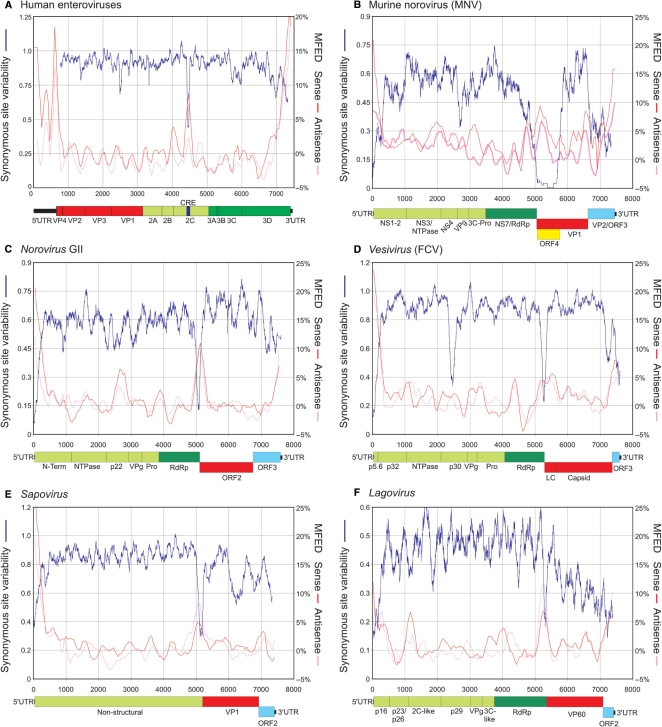
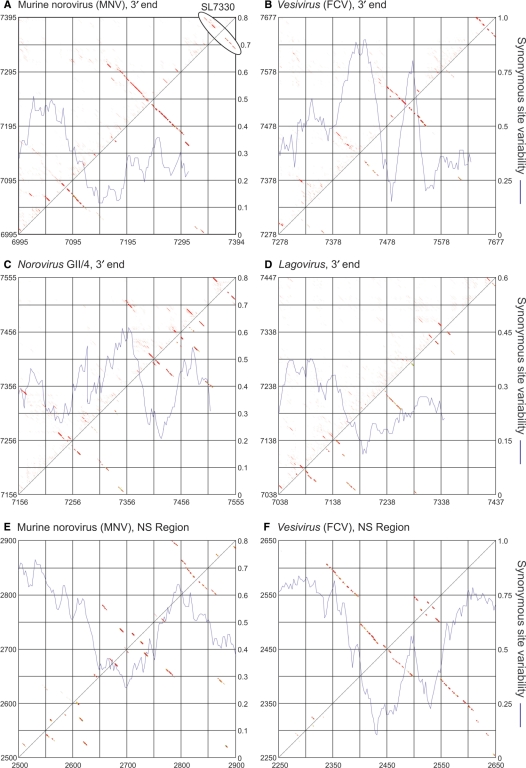
The mechanism and role of RNA structure elements in the replication and translation of Caliciviridae remains poorly understood. Several algorithmically independent methods were used to predict secondary structures within the Norovirus, Sapovirus, Vesivirus and Lagovirus genera. All showed profound suppression of synonymous site variability (SSSV) at genomic 5' ends and the start of the sub-genomic (sg) transcript, consistent with evolutionary constraints from underlying RNA structure. A newly developed thermodynamic scanning method predicted RNA folding mapping precisely to regions of SSSV and at the genomic 3' end. These regions contained several evolutionarily conserved RNA secondary structures, of variable size and positions. However, all caliciviruses contained 3' terminal hairpins, and stem-loops in the anti-genomic strand invariably six bases upstream of the sg transcript, indicating putative roles as sg promoters. Using the murine norovirus (MNV) reverse-genetics system, disruption of 5' end stem-loops produced approximately 15- to 20-fold infectivity reductions, while disruption of the RNA structure in the sg promoter region and at the 3' end entirely destroyed replication ability. Restoration of infectivity by repair mutations in the sg promoter region confirmed a functional role for the RNA secondary structure, not the sequence. This study provides comprehensive bioinformatic resources for future functional studies of MNV and other caliciviruses.
Figures





Similar articles
-
Functions of the 5' and 3' ends of calicivirus genomes.Virus Res. 2015 Aug 3;206:134-43. doi: 10.1016/j.virusres.2015.02.002. Epub 2015 Feb 9. Virus Res. 2015. PMID: 25678268 Free PMC article. Review.
-
The murine norovirus core subgenomic RNA promoter consists of a stable stem-loop that can direct accurate initiation of RNA synthesis.J Virol. 2015 Jan 15;89(2):1218-29. doi: 10.1128/JVI.02432-14. Epub 2014 Nov 12. J Virol. 2015. PMID: 25392209 Free PMC article.
-
Inter- and intragenus structural variations in caliciviruses and their functional implications.J Virol. 2004 Jun;78(12):6469-79. doi: 10.1128/JVI.78.12.6469-6479.2004. J Virol. 2004. PMID: 15163740 Free PMC article.
-
Functional analysis of RNA structures present at the 3' extremity of the murine norovirus genome: the variable polypyrimidine tract plays a role in viral virulence.J Virol. 2010 Mar;84(6):2859-70. doi: 10.1128/JVI.02053-09. Epub 2010 Jan 6. J Virol. 2010. PMID: 20053745 Free PMC article.
-
Caliciviridae Other Than Noroviruses.Viruses. 2019 Mar 21;11(3):286. doi: 10.3390/v11030286. Viruses. 2019. PMID: 30901945 Free PMC article. Review.
Cited by
-
Functions of the 5' and 3' ends of calicivirus genomes.Virus Res. 2015 Aug 3;206:134-43. doi: 10.1016/j.virusres.2015.02.002. Epub 2015 Feb 9. Virus Res. 2015. PMID: 25678268 Free PMC article. Review.
-
A conserved predicted pseudoknot in the NS2A-encoding sequence of West Nile and Japanese encephalitis flaviviruses suggests NS1' may derive from ribosomal frameshifting.Virol J. 2009 Feb 5;6:14. doi: 10.1186/1743-422X-6-14. Virol J. 2009. PMID: 19196463 Free PMC article.
-
Discovery of a small arterivirus gene that overlaps the GP5 coding sequence and is important for virus production.J Gen Virol. 2011 May;92(Pt 5):1097-1106. doi: 10.1099/vir.0.029264-0. Epub 2011 Feb 9. J Gen Virol. 2011. PMID: 21307223 Free PMC article.
-
The dinucleotide composition of the Zika virus genome is shaped by conflicting evolutionary pressures in mammalian hosts and mosquito vectors.PLoS Biol. 2021 Apr 19;19(4):e3001201. doi: 10.1371/journal.pbio.3001201. eCollection 2021 Apr. PLoS Biol. 2021. PMID: 33872300 Free PMC article.
-
Recombination at the emergence of the pathogenic rabbit haemorrhagic disease virus Lagovirus europaeus/GI.2.Sci Rep. 2020 Sep 2;10(1):14502. doi: 10.1038/s41598-020-71303-4. Sci Rep. 2020. PMID: 32879332 Free PMC article.
