

Protein disulfide isomerase acts as an injury response signal that enhances fibrin generation via tissue factor activation
- PMID: 18274674
- PMCID: PMC2242616
- DOI: 10.1172/JCI32376
Protein disulfide isomerase acts as an injury response signal that enhances fibrin generation via tissue factor activation
Abstract
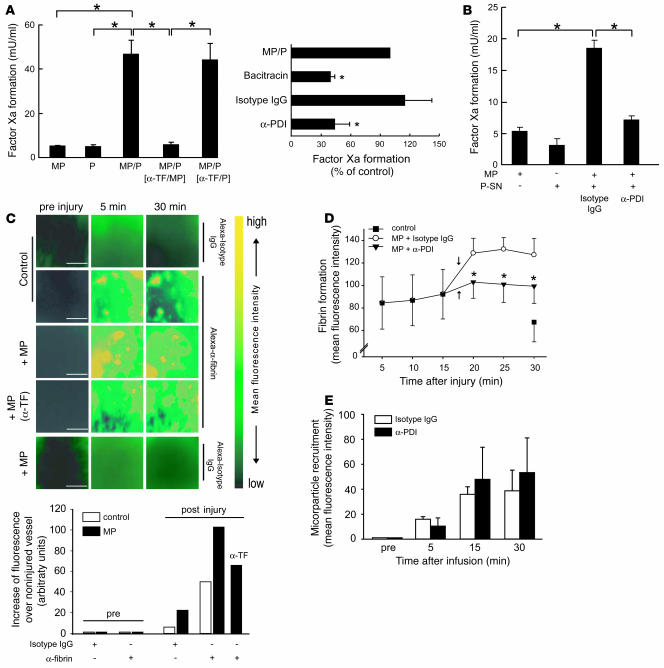
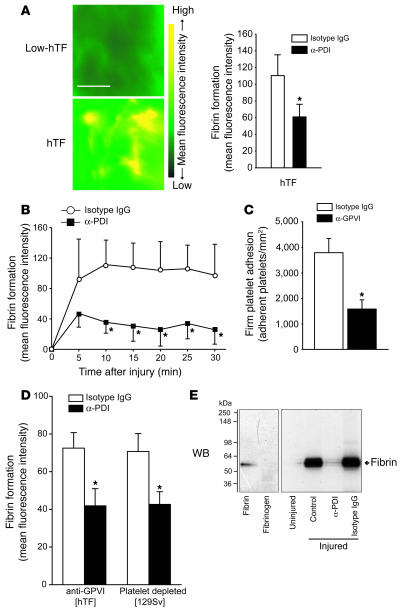
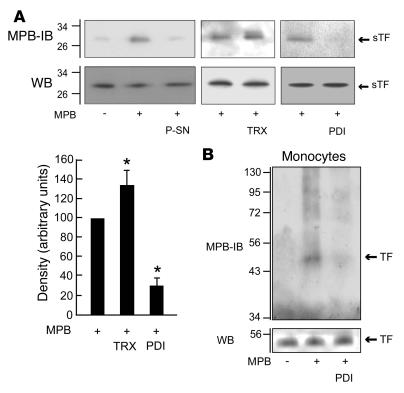
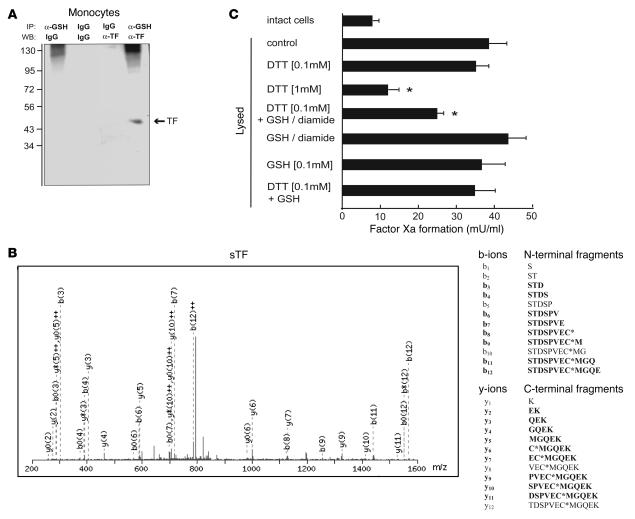
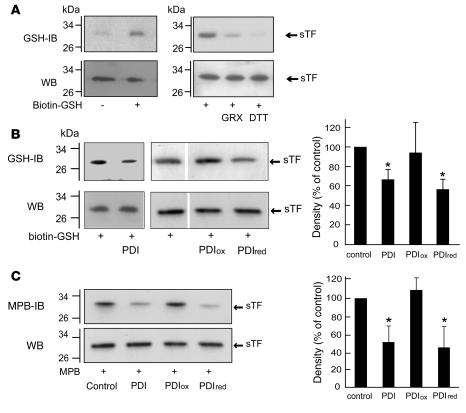
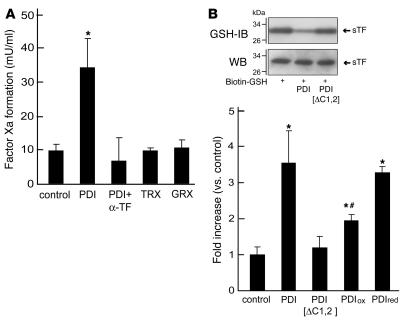
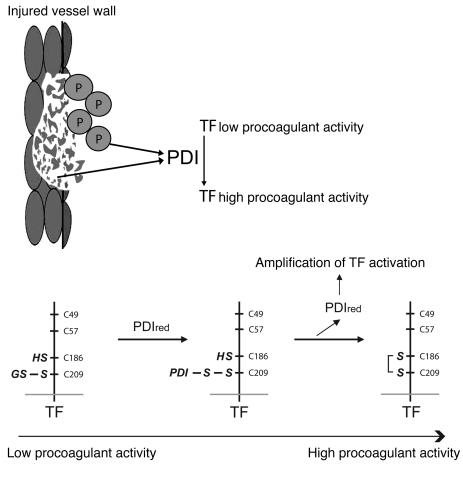
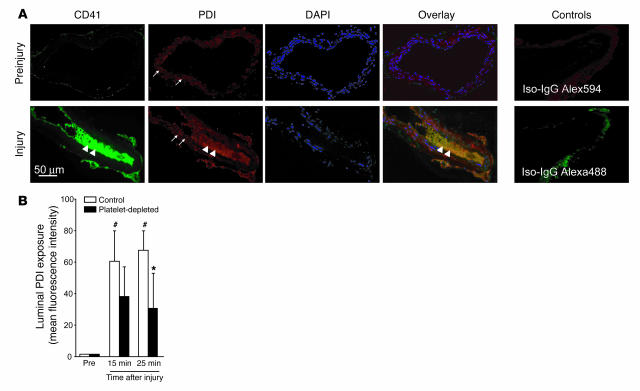
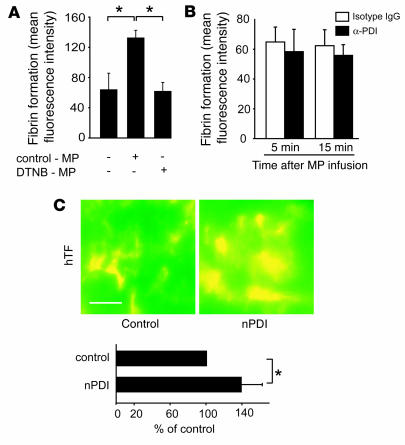
The activation of initiator protein tissue factor (TF) is likely to be a crucial step in the blood coagulation process, which leads to fibrin formation. The stimuli responsible for inducing TF activation are largely undefined. Here we show that the oxidoreductase protein disulfide isomerase (PDI) directly promotes TF-dependent fibrin production during thrombus formation in vivo. After endothelial denudation of mouse carotid arteries, PDI was released at the injury site from adherent platelets and disrupted vessel wall cells. Inhibition of PDI decreased TF-triggered fibrin formation in different in vivo murine models of thrombus formation, as determined by intravital fluorescence microscopy. PDI infusion increased - and, under conditions of decreased platelet adhesion, PDI inhibition reduced - fibrin generation at the injury site, indicating that PDI can directly initiate blood coagulation. In vitro, human platelet-secreted PDI contributed to the activation of cryptic TF on microvesicles (microparticles). Mass spectrometry analyses indicated that part of the extracellular cysteine 209 of TF was constitutively glutathionylated. Mixed disulfide formation contributed to maintaining TF in a state of low functionality. We propose that reduced PDI activates TF by isomerization of a mixed disulfide and a free thiol to an intramolecular disulfide. Our findings suggest that disulfide isomerases can act as injury response signals that trigger the activation of fibrin formation following vessel injury.
Figures
Similar articles
-
Protein disulfide isomerase as a trigger for tissue factor-dependent fibrin generation.Thromb Res. 2008;122 Suppl 1:S19-22. doi: 10.1016/S0049-3848(08)70013-6. Thromb Res. 2008. PMID: 18691493 Review.
-
A critical role for extracellular protein disulfide isomerase during thrombus formation in mice.J Clin Invest. 2008 Mar;118(3):1123-31. doi: 10.1172/JCI34134. J Clin Invest. 2008. PMID: 18292814 Free PMC article.
-
Tissue factor coagulant function is enhanced by protein-disulfide isomerase independent of oxidoreductase activity.J Biol Chem. 2007 Aug 31;282(35):25416-24. doi: 10.1074/jbc.M702410200. Epub 2007 Jul 5. J Biol Chem. 2007. PMID: 17613528
-
Extracellular protein disulfide isomerase regulates feedback activation of platelet thrombin generation via modulation of coagulation factor binding.J Thromb Haemost. 2011 Nov;9(11):2278-90. doi: 10.1111/j.1538-7836.2011.04509.x. J Thromb Haemost. 2011. PMID: 21929690
-
Regulatory role of thiol isomerases in thrombus formation.Expert Rev Hematol. 2018 May;11(5):437-448. doi: 10.1080/17474086.2018.1452612. Epub 2018 Mar 28. Expert Rev Hematol. 2018. PMID: 29542339 Free PMC article. Review.
Cited by
-
Levels of peripheral blood routine, biochemical and coagulation parameters in patients with hemorrhagic fever with renal syndrome and their relationship with prognosis: an observational cohort study.BMC Infect Dis. 2024 Jan 11;24(1):75. doi: 10.1186/s12879-023-08777-w. BMC Infect Dis. 2024. PMID: 38212688 Free PMC article.
-
Platelet protein disulfide isomerase is required for thrombus formation but not for hemostasis in mice.Blood. 2013 Aug 8;122(6):1052-61. doi: 10.1182/blood-2013-03-492504. Epub 2013 Jun 20. Blood. 2013. PMID: 23788140 Free PMC article.
-
Thromboinflammation in Sepsis and Heparin: A Review of Literature and Pathophysiology.In Vivo. 2022 Nov-Dec;36(6):2542-2557. doi: 10.21873/invivo.12991. In Vivo. 2022. PMID: 36309378 Free PMC article. Review.
-
The Human Platelet as an Innate Immune Cell: Interactions Between Activated Platelets and the Complement System.Front Immunol. 2019 Jul 10;10:1590. doi: 10.3389/fimmu.2019.01590. eCollection 2019. Front Immunol. 2019. PMID: 31354729 Free PMC article. Review.
-
Thiol isomerases in thrombus formation.Circ Res. 2014 Mar 28;114(7):1162-73. doi: 10.1161/CIRCRESAHA.114.301808. Circ Res. 2014. PMID: 24677236 Free PMC article. Review.
References
-
- Dahlback B. Blood coagulation. Lancet. 2000;355:1627–1632. - PubMed
-
- Schenone M., Furie B.C., Furie B. The blood coagulation cascade. Curr. Opin. Hematol. 2004;11:272–277. - PubMed
-
- Gomez K., McVey K.H. Tissue factor initiated blood coagulation. Front. Biosci. 2006;11:1349–1359. - PubMed
-
- Mackman N., Tilley R.E., Key N.S. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler. Thromb. Vasc. Biol. . 2007;27:1687–1693. - PubMed
-
- Steffel J., Lüscher T.F., Tanner F.C. Tissue factor in cardiovascular diseases: molecular mechanisms and clinical implications. Circulation. . 2006;113:722–731. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
