

Transcription factors bind thousands of active and inactive regions in the Drosophila blastoderm
- PMID: 18271625
- PMCID: PMC2235902
- DOI: 10.1371/journal.pbio.0060027
Transcription factors bind thousands of active and inactive regions in the Drosophila blastoderm
Abstract
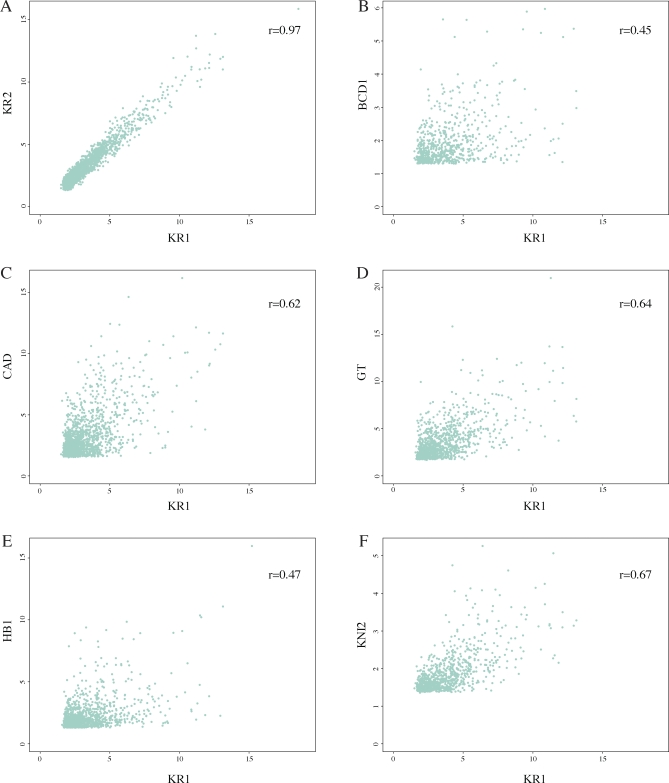
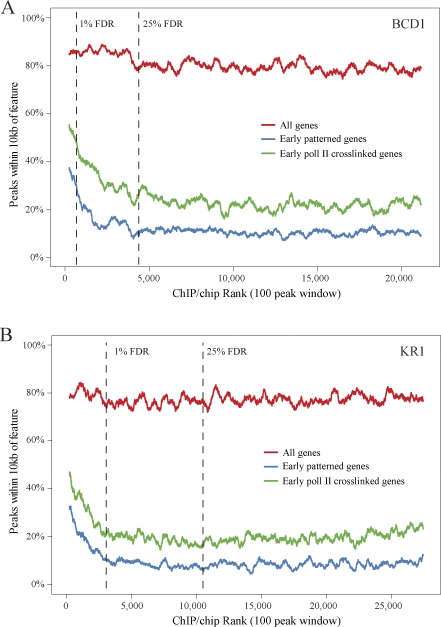
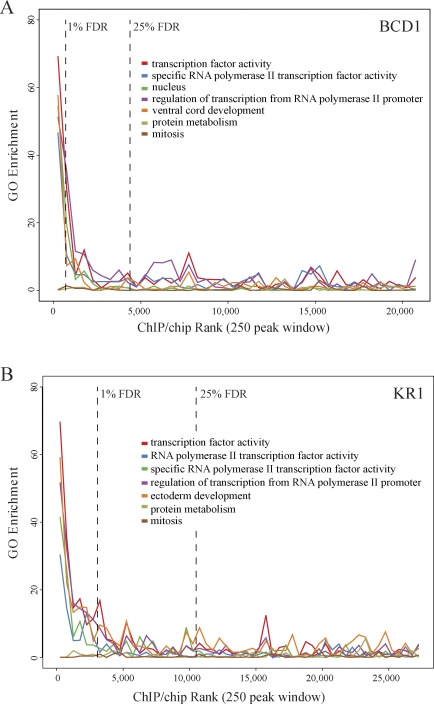
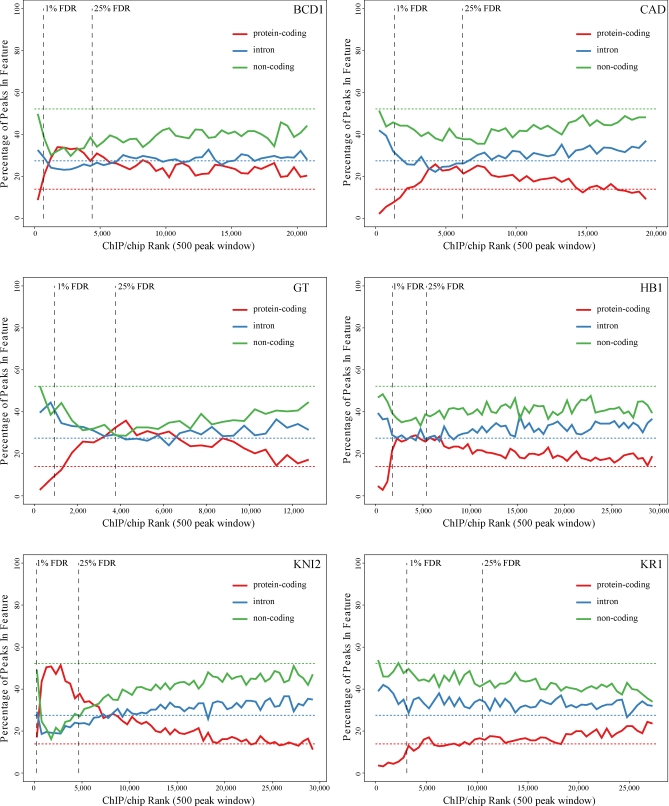
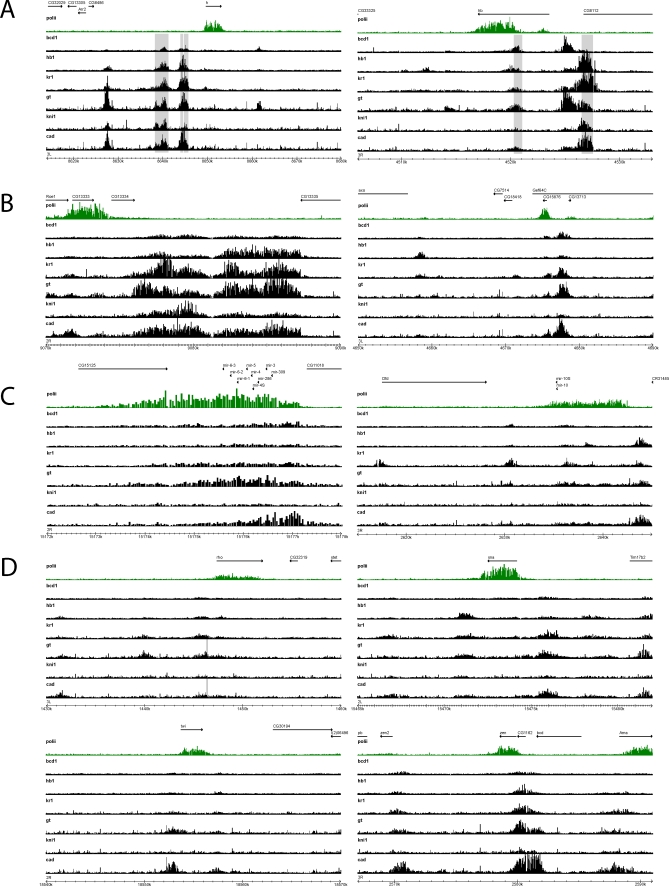
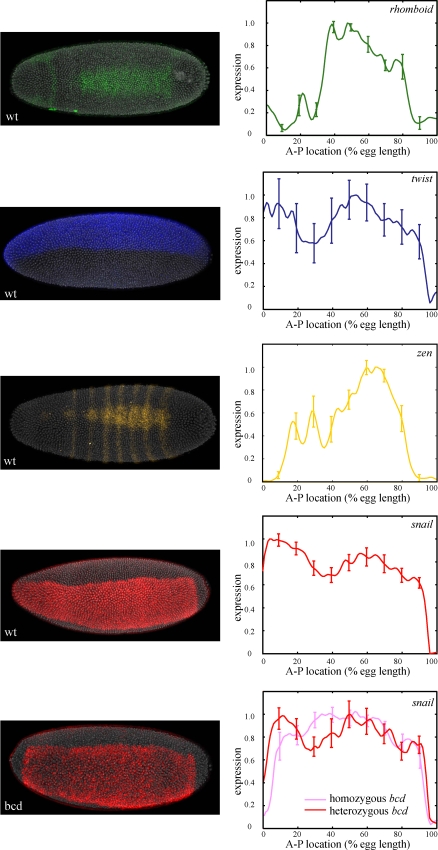
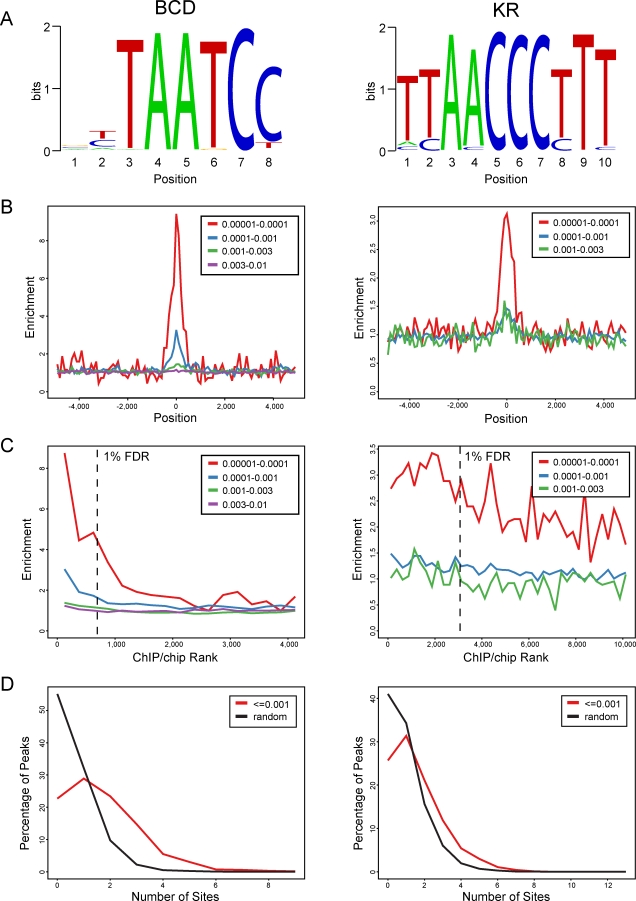
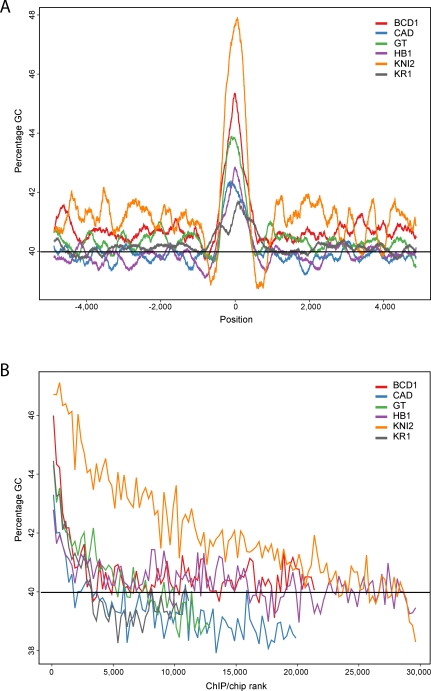
Identifying the genomic regions bound by sequence-specific regulatory factors is central both to deciphering the complex DNA cis-regulatory code that controls transcription in metazoans and to determining the range of genes that shape animal morphogenesis. We used whole-genome tiling arrays to map sequences bound in Drosophila melanogaster embryos by the six maternal and gap transcription factors that initiate anterior-posterior patterning. We find that these sequence-specific DNA binding proteins bind with quantitatively different specificities to highly overlapping sets of several thousand genomic regions in blastoderm embryos. Specific high- and moderate-affinity in vitro recognition sequences for each factor are enriched in bound regions. This enrichment, however, is not sufficient to explain the pattern of binding in vivo and varies in a context-dependent manner, demonstrating that higher-order rules must govern targeting of transcription factors. The more highly bound regions include all of the over 40 well-characterized enhancers known to respond to these factors as well as several hundred putative new cis-regulatory modules clustered near developmental regulators and other genes with patterned expression at this stage of embryogenesis. The new targets include most of the microRNAs (miRNAs) transcribed in the blastoderm, as well as all major zygotically transcribed dorsal-ventral patterning genes, whose expression we show to be quantitatively modulated by anterior-posterior factors. In addition to these highly bound regions, there are several thousand regions that are reproducibly bound at lower levels. However, these poorly bound regions are, collectively, far more distant from genes transcribed in the blastoderm than highly bound regions; are preferentially found in protein-coding sequences; and are less conserved than highly bound regions. Together these observations suggest that many of these poorly bound regions are not involved in early-embryonic transcriptional regulation, and a significant proportion may be nonfunctional. Surprisingly, for five of the six factors, their recognition sites are not unambiguously more constrained evolutionarily than the immediate flanking DNA, even in more highly bound and presumably functional regions, indicating that comparative DNA sequence analysis is limited in its ability to identify functional transcription factor targets.
Conflict of interest statement
Figures

Similar articles
-
Developmental roles of 21 Drosophila transcription factors are determined by quantitative differences in binding to an overlapping set of thousands of genomic regions.Genome Biol. 2009;10(7):R80. doi: 10.1186/gb-2009-10-7-r80. Epub 2009 Jul 23. Genome Biol. 2009. PMID: 19627575 Free PMC article.
-
DNA regions bound at low occupancy by transcription factors do not drive patterned reporter gene expression in Drosophila.Proc Natl Acad Sci U S A. 2012 Dec 26;109(52):21330-5. doi: 10.1073/pnas.1209589110. Epub 2012 Dec 10. Proc Natl Acad Sci U S A. 2012. PMID: 23236164 Free PMC article.
-
Zelda binding in the early Drosophila melanogaster embryo marks regions subsequently activated at the maternal-to-zygotic transition.PLoS Genet. 2011 Oct;7(10):e1002266. doi: 10.1371/journal.pgen.1002266. Epub 2011 Oct 20. PLoS Genet. 2011. PMID: 22028662 Free PMC article.
-
Building quantitative, three-dimensional atlases of gene expression and morphology at cellular resolution.Wiley Interdiscip Rev Dev Biol. 2013 Nov-Dec;2(6):767-79. doi: 10.1002/wdev.107. Epub 2013 Feb 4. Wiley Interdiscip Rev Dev Biol. 2013. PMID: 24123936 Free PMC article. Review.
-
A census of human transcription factors: function, expression and evolution.Nat Rev Genet. 2009 Apr;10(4):252-63. doi: 10.1038/nrg2538. Nat Rev Genet. 2009. PMID: 19274049 Review.
Cited by
-
Natural variation in the regulation of neurodevelopmental genes modifies flight performance in Drosophila.PLoS Genet. 2021 Mar 18;17(3):e1008887. doi: 10.1371/journal.pgen.1008887. eCollection 2021 Mar. PLoS Genet. 2021. PMID: 33735180 Free PMC article.
-
Insights from genomic profiling of transcription factors.Nat Rev Genet. 2009 Sep;10(9):605-16. doi: 10.1038/nrg2636. Epub 2009 Aug 11. Nat Rev Genet. 2009. PMID: 19668247 Free PMC article. Review.
-
NagRBt Is a Pleiotropic and Dual Transcriptional Regulator in Bacillus thuringiensis.Front Microbiol. 2018 Sep 11;9:1899. doi: 10.3389/fmicb.2018.01899. eCollection 2018. Front Microbiol. 2018. PMID: 30254611 Free PMC article.
-
Nonparametric identification of regulatory interactions from spatial and temporal gene expression data.BMC Bioinformatics. 2010 Aug 4;11:413. doi: 10.1186/1471-2105-11-413. BMC Bioinformatics. 2010. PMID: 20684787 Free PMC article.
-
Regulation of the histone demethylase JMJD1A by hypoxia-inducible factor 1 alpha enhances hypoxic gene expression and tumor growth.Mol Cell Biol. 2010 Jan;30(1):344-53. doi: 10.1128/MCB.00444-09. Mol Cell Biol. 2010. PMID: 19858293 Free PMC article.
References
-
- Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, et al. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–2195. - PubMed
-
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, et al. The sequence of the human genome. Science. 2001;291:1304–1351. - PubMed
-
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. - PubMed
-
- Biggin MD, Tjian R. Transcriptional regulation in Drosophila: the post-genome challenge. Funct Integr Genomics. 2001;1:223–234. - PubMed
-
- Davidson EH. Genomic regulatory systems: development and evolution. San Diego: Academic Press; 2001. 261
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
