

SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model
- PMID: 18219391
- PMCID: PMC2213375
- DOI: 10.1172/JCI34060
SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model
Abstract
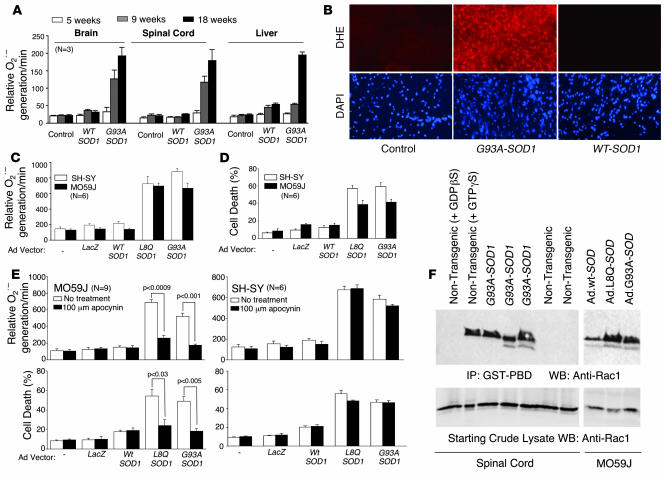
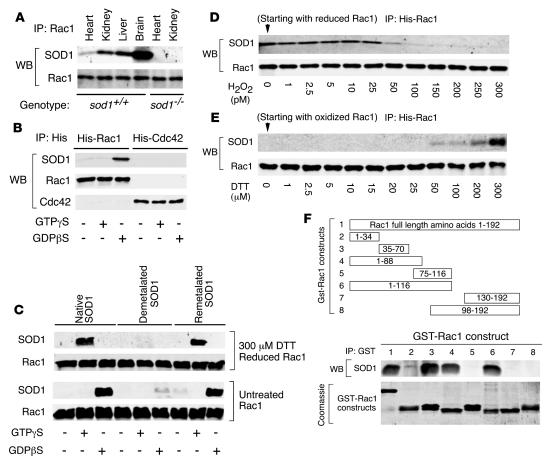
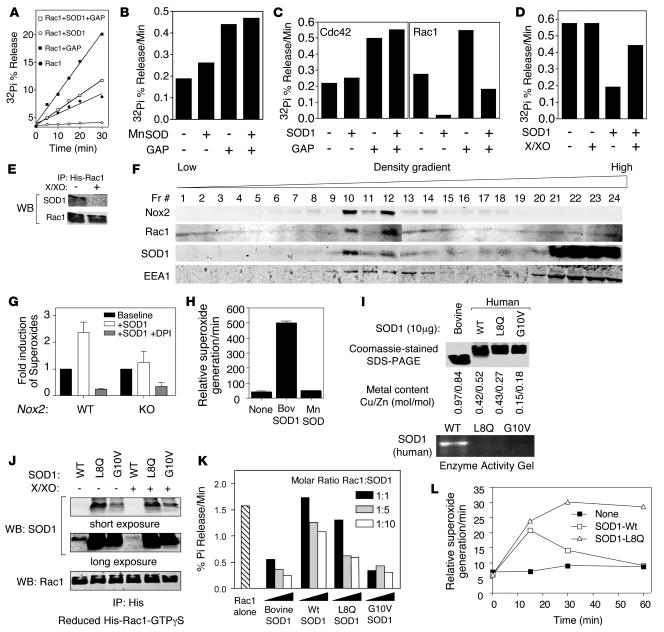
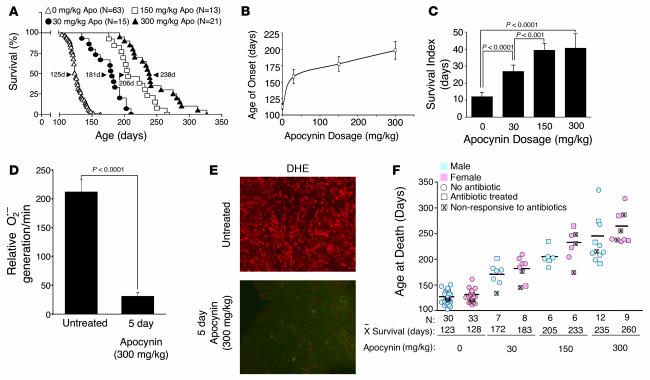
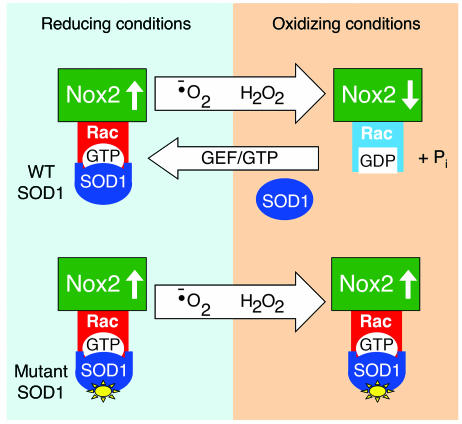
Neurodegeneration in familial amyotrophic lateral sclerosis (ALS) is associated with enhanced redox stress caused by dominant mutations in superoxide dismutase-1 (SOD1). SOD1 is a cytosolic enzyme that facilitates the conversion of superoxide (O(2)(*-)) to H(2)O(2). Here we demonstrate that SOD1 is not just a catabolic enzyme, but can also directly regulate NADPH oxidase-dependent (Nox-dependent) O(2)(*-) production by binding Rac1 and inhibiting its GTPase activity. Oxidation of Rac1 by H(2)O(2) uncoupled SOD1 binding in a reversible fashion, producing a self-regulating redox sensor for Nox-derived O(2)(*-) production. This process of redox-sensitive uncoupling of SOD1 from Rac1 was defective in SOD1 ALS mutants, leading to enhanced Rac1/Nox activation in transgenic mouse tissues and cell lines expressing ALS SOD1 mutants. Glial cell toxicity associated with expression of SOD1 mutants in culture was significantly attenuated by treatment with the Nox inhibitor apocynin. Treatment of ALS mice with apocynin also significantly increased their average life span. This redox sensor mechanism may explain the gain-of-function seen with certain SOD1 mutations associated with ALS and defines new therapeutic targets.
Figures
Comment in
-
Revisiting oxidative damage in ALS: microglia, Nox, and mutant SOD1.J Clin Invest. 2008 Feb;118(2):474-8. doi: 10.1172/JCI34613. J Clin Invest. 2008. PMID: 18219386 Free PMC article.
Similar articles
-
Alsin and SOD1(G93A) proteins regulate endosomal reactive oxygen species production by glial cells and proinflammatory pathways responsible for neurotoxicity.J Biol Chem. 2011 Nov 18;286(46):40151-62. doi: 10.1074/jbc.M111.279711. Epub 2011 Sep 20. J Biol Chem. 2011. PMID: 21937428 Free PMC article.
-
Revisiting oxidative damage in ALS: microglia, Nox, and mutant SOD1.J Clin Invest. 2008 Feb;118(2):474-8. doi: 10.1172/JCI34613. J Clin Invest. 2008. PMID: 18219386 Free PMC article.
-
The NADPH oxidase pathway is dysregulated by the P2X7 receptor in the SOD1-G93A microglia model of amyotrophic lateral sclerosis.J Immunol. 2013 May 15;190(10):5187-95. doi: 10.4049/jimmunol.1203262. Epub 2013 Apr 15. J Immunol. 2013. PMID: 23589615
-
Transgenic mouse model for familial amyotrophic lateral sclerosis with superoxide dismutase-1 mutation.Neuropathology. 2001 Mar;21(1):82-92. doi: 10.1046/j.1440-1789.2001.00361.x. Neuropathology. 2001. PMID: 11304046 Review.
-
Redox modifier genes and pathways in amyotrophic lateral sclerosis.Antioxid Redox Signal. 2009 Jul;11(7):1569-86. doi: 10.1089/ars.2008.2414. Antioxid Redox Signal. 2009. PMID: 19187001 Free PMC article. Review.
Cited by
-
Nuclear Localization of Human SOD1 in Motor Neurons in Mouse Model and Patient Amyotrophic Lateral Sclerosis: Possible Links to Cholinergic Phenotype, NADPH Oxidase, Oxidative Stress, and DNA Damage.Int J Mol Sci. 2024 Aug 22;25(16):9106. doi: 10.3390/ijms25169106. Int J Mol Sci. 2024. PMID: 39201793 Free PMC article.
-
Oxidative Cysteine Post Translational Modifications Drive the Redox Code Underlying Neurodegeneration and Amyotrophic Lateral Sclerosis.Antioxidants (Basel). 2024 Jul 23;13(8):883. doi: 10.3390/antiox13080883. Antioxidants (Basel). 2024. PMID: 39199129 Free PMC article. Review.
-
The prion-like effect and prion-like protein targeting strategy in amyotrophic lateral sclerosis.Heliyon. 2024 Jul 22;10(15):e34963. doi: 10.1016/j.heliyon.2024.e34963. eCollection 2024 Aug 15. Heliyon. 2024. PMID: 39170125 Free PMC article. Review.
-
Exploring antioxidant strategies in the pathogenesis of ALS.Open Life Sci. 2024 Mar 28;19(1):20220842. doi: 10.1515/biol-2022-0842. eCollection 2024. Open Life Sci. 2024. PMID: 38585631 Free PMC article. Review.
-
New Insights into Oxidative Stress and Inflammatory Response in Neurodegenerative Diseases.Int J Mol Sci. 2024 Feb 26;25(5):2698. doi: 10.3390/ijms25052698. Int J Mol Sci. 2024. PMID: 38473944 Free PMC article. Review.
References
-
- McCord J.M., Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969;244:6049–6055. - PubMed
-
- Rosen D.R., et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. - PubMed
-
- Bruijn L.I., Miller T.M., Cleveland D.W. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu. Rev. Neurosci. 2004;27:723–749. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
