

Activation of peroxisome proliferator-activated receptor gamma (PPARgamma) suppresses Rho GTPases in human brain microvascular endothelial cells and inhibits adhesion and transendothelial migration of HIV-1 infected monocytes
- PMID: 18209083
- PMCID: PMC3384722
- DOI: 10.4049/jimmunol.180.3.1854
Activation of peroxisome proliferator-activated receptor gamma (PPARgamma) suppresses Rho GTPases in human brain microvascular endothelial cells and inhibits adhesion and transendothelial migration of HIV-1 infected monocytes
Abstract
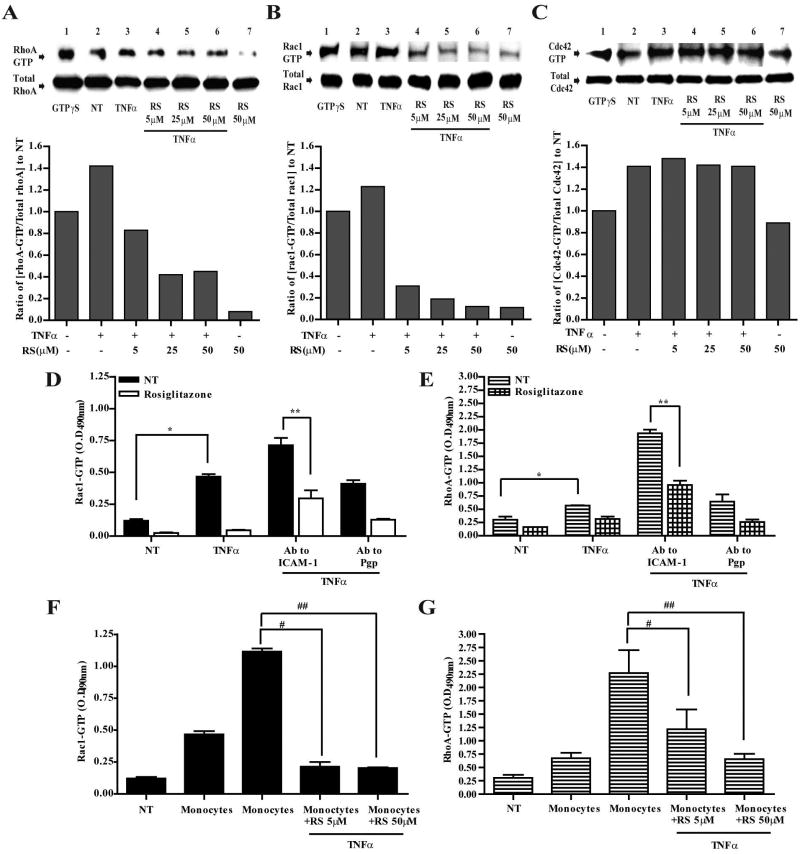
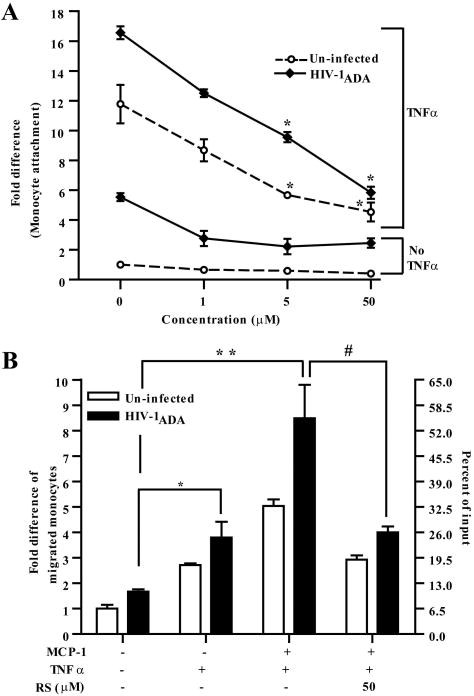
Under inflammatory conditions (including HIV-1 encephalitis and multiple sclerosis), activated brain endothelium enhances the adhesion and transmigration of monocytes across the blood-brain barrier (BBB). Synthetic ligands that activate the peroxisome proliferator-activated receptors (PPARs) have anti-inflammatory properties, and PPAR stimulation prevents the interaction of leukocytes with cytokine stimulated-endothelium. However, the mechanism underlying these effects of PPAR ligands and their ability to intervene with leukocyte adhesion and migration across brain endothelial cells has yet to be explored. For the first time, using primary human brain endothelial cells (BMVEC), we demonstrated that monocyte adhesion and transendothelial migration across inflamed endothelium were markedly reduced by PPARgamma activation. In contrast to non-brain-derived endothelial cells, PPARalpha activation in the BMVEC had no significant effect on monocyte-endothelial interaction. Previously, our work indicated a critical role of Rho GTPases (like RhoA) in BMVEC to control migration of HIV-1 infected monocytes across BBB. In this study, we show that in the BMVEC PPARgamma stimulation prevented activation of two GTPases, Rac1 and RhoA, which correlated with decreased monocyte adhesion to and migration across brain endothelium. Relevant to HIV-1 neuropathogenesis, enhanced adhesion and migration of HIV-1 infected monocytes across the BBB were significantly reduced when BMVEC were treated with PPARgamma agonist. These findings indicate that Rac1 and RhoA inhibition by PPARgamma agonists could be a new approach for treatment of neuroinflammation by preventing monocyte migration across the BBB.
Conflict of interest statement
The authors have no financial conflict of interest.
Figures





Similar articles
-
Glycogen synthase kinase 3β inhibition prevents monocyte migration across brain endothelial cells via Rac1-GTPase suppression and down-regulation of active integrin conformation.Am J Pathol. 2012 Oct;181(4):1414-25. doi: 10.1016/j.ajpath.2012.06.018. Epub 2012 Aug 3. Am J Pathol. 2012. PMID: 22863953 Free PMC article.
-
Rho-mediated regulation of tight junctions during monocyte migration across the blood-brain barrier in HIV-1 encephalitis (HIVE).Blood. 2006 Jun 15;107(12):4770-80. doi: 10.1182/blood-2005-11-4721. Epub 2006 Feb 14. Blood. 2006. PMID: 16478881 Free PMC article.
-
Mechanisms for the transendothelial migration of HIV-1-infected monocytes into brain.J Immunol. 1996 Feb 1;156(3):1284-95. J Immunol. 1996. PMID: 8558009
-
Interactions between macrophages and brain microvascular endothelial cells: role in pathogenesis of HIV-1 infection and blood - brain barrier function.J Neurovirol. 1999 Dec;5(6):659-69. doi: 10.3109/13550289909021294. J Neurovirol. 1999. PMID: 10602406 Review.
-
Model systems for studies of leukocyte migration across the blood - brain barrier.J Neurovirol. 1999 Dec;5(6):579-90. doi: 10.3109/13550289909021287. J Neurovirol. 1999. PMID: 10602399 Review.
Cited by
-
Successful isolation of infectious and high titer human monocyte-derived HIV-1 from two subjects with discontinued therapy.PLoS One. 2013 May 31;8(5):e65071. doi: 10.1371/journal.pone.0065071. Print 2013. PLoS One. 2013. PMID: 23741458 Free PMC article.
-
Glycogen synthase kinase 3β inhibition prevents monocyte migration across brain endothelial cells via Rac1-GTPase suppression and down-regulation of active integrin conformation.Am J Pathol. 2012 Oct;181(4):1414-25. doi: 10.1016/j.ajpath.2012.06.018. Epub 2012 Aug 3. Am J Pathol. 2012. PMID: 22863953 Free PMC article.
-
FXR protects lung from lipopolysaccharide-induced acute injury.Mol Endocrinol. 2012 Jan;26(1):27-36. doi: 10.1210/me.2011-0042. Epub 2011 Dec 1. Mol Endocrinol. 2012. PMID: 22135065 Free PMC article.
-
The synthetic cannabinoid R(+)WIN55,212-2 augments interferon-β expression via peroxisome proliferator-activated receptor-α.J Biol Chem. 2012 Jul 20;287(30):25440-53. doi: 10.1074/jbc.M112.371757. Epub 2012 May 31. J Biol Chem. 2012. PMID: 22654113 Free PMC article.
-
Establishment of primary cultures of human brain microvascular endothelial cells to provide an in vitro cellular model of the blood-brain barrier.Nat Protoc. 2010 Jul;5(7):1265-72. doi: 10.1038/nprot.2010.76. Epub 2010 Jun 10. Nat Protoc. 2010. PMID: 20595955 Free PMC article.
References
-
- Engelhardt B, Ransohoff RM. The ins and outs of T-lymphocyte trafficking to the CNS: anatomical sites and molecular mechanisms. Trends Immunol. 2005;26:485–495. - PubMed
-
- Kanwar JR. Anti-inflammatory immunotherapy for multiple sclerosis/experimental autoimmune encephalomyelitis (EAE) disease. Curr Med Chem. 2005;12:2947–2962. - PubMed
-
- Persidsky Y, Gendelman HE. Mononuclear phagocyte immunity and the neuropathogenesis of HIV-1 infection. J Leukoc Biol. 2003;74:691–701. - PubMed
-
- Eugenin EA, Osiecki K, Lopez L, Goldstein H, Calderon TM, Berman JW. CCL2/monocyte chemoattractant protein-1 mediates enhanced transmigration of human immunodeficiency virus (HIV)-infected leukocytes across the blood-brain barrier: a potential mechanism of HIV-CNS invasion and NeuroAIDS. J Neurosci. 2006;26:1098–1106. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials