

Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion
- PMID: 18077707
- PMCID: PMC2258938
- DOI: 10.1128/JVI.02053-07
Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion
Abstract
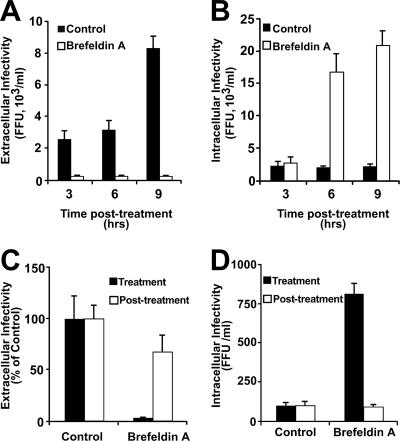
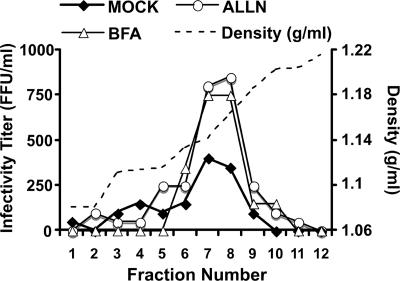
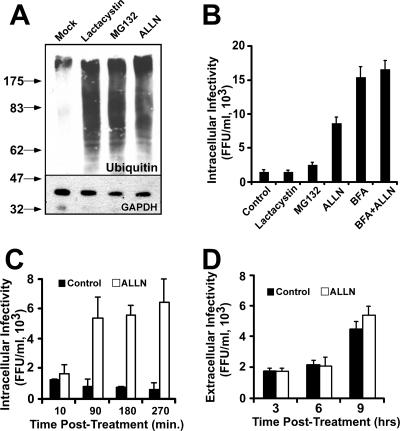
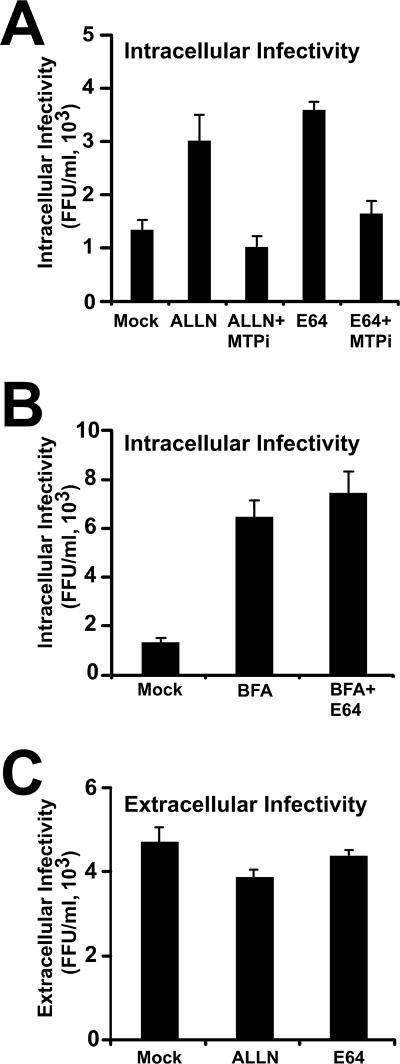
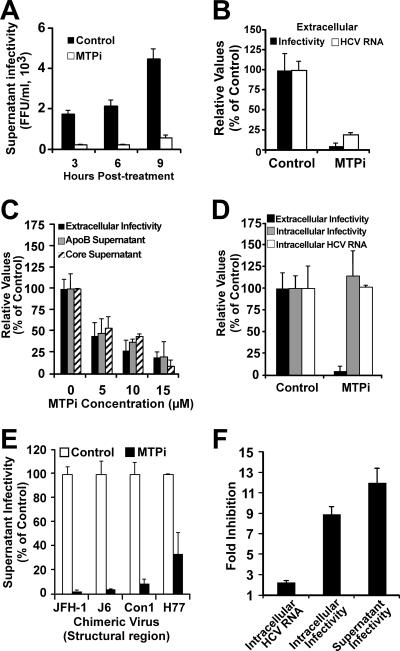
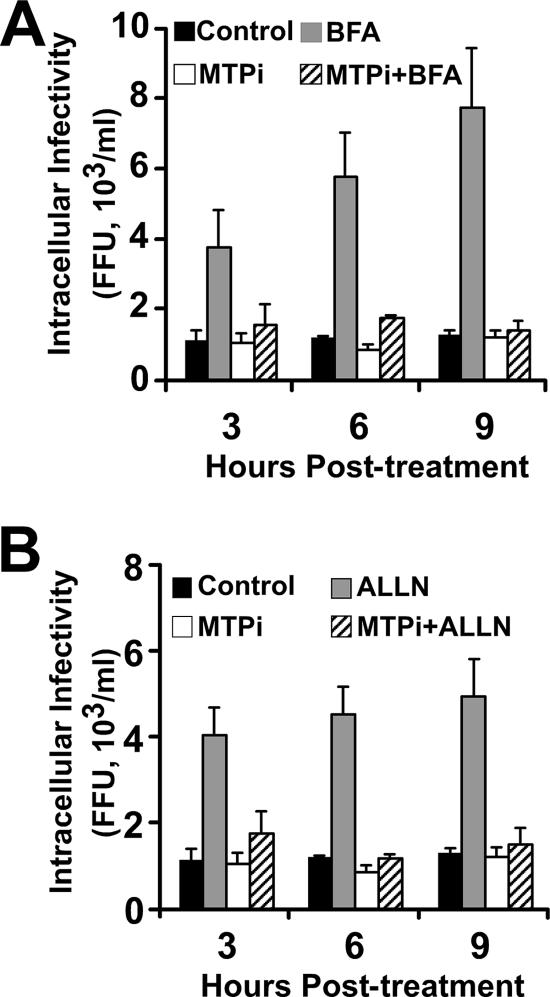
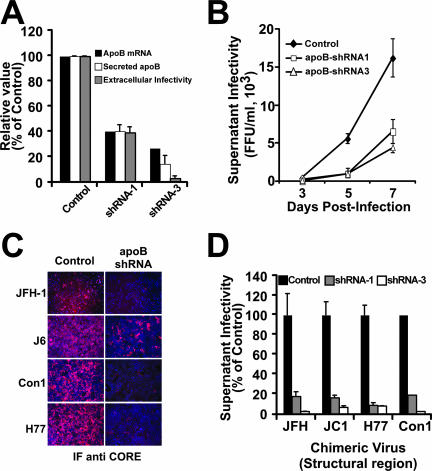
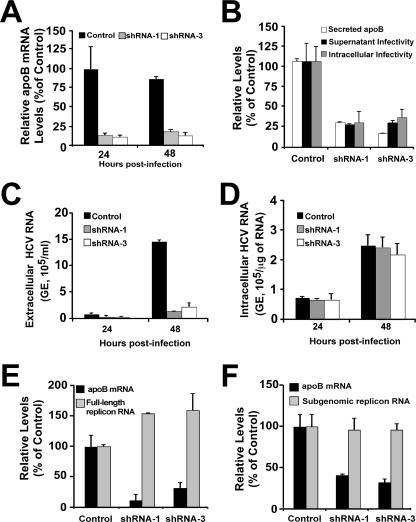
Intracellular infectious hepatitis C virus (HCV) particles display a distinctly higher buoyant density than do secreted virus particles, suggesting that the characteristic low density of extracellular HCV particles is acquired during viral egress. We took advantage of this difference to examine the determinants of assembly, maturation, degradation, and egress of infectious HCV particles. The results demonstrate that HCV assembly and maturation occur in the endoplasmic reticulum (ER) and post-ER compartments, respectively, and that both depend on microsomal transfer protein and apolipoprotein B, in a manner that parallels the formation of very-low-density lipoproteins (VLDL). In addition, they illustrate that only low-density particles are efficiently secreted and that immature particles are actively degraded, in a proteasome-independent manner, in a post-ER compartment of the cell. These results suggest that by coopting the VLDL assembly, maturation, degradation, and secretory machinery of the cell, HCV acquires its hepatocyte tropism and, by mimicry, its tendency to persist.
Figures
Similar articles
-
Hepatitis C Virus Lipoviroparticles Assemble in the Endoplasmic Reticulum (ER) and Bud off from the ER to the Golgi Compartment in COPII Vesicles.J Virol. 2017 Jul 12;91(15):e00499-17. doi: 10.1128/JVI.00499-17. Print 2017 Aug 1. J Virol. 2017. PMID: 28515296 Free PMC article.
-
Apolipoprotein E likely contributes to a maturation step of infectious hepatitis C virus particles and interacts with viral envelope glycoproteins.J Virol. 2014 Nov;88(21):12422-37. doi: 10.1128/JVI.01660-14. Epub 2014 Aug 13. J Virol. 2014. PMID: 25122793 Free PMC article.
-
Apolipoprotein E, but Not Apolipoprotein B, Is Essential for Efficient Cell-to-Cell Transmission of Hepatitis C Virus.J Virol. 2015 Oct;89(19):9962-73. doi: 10.1128/JVI.00577-15. Epub 2015 Jul 22. J Virol. 2015. PMID: 26202245 Free PMC article.
-
Hepatitis C virus particles and lipoprotein metabolism.Semin Liver Dis. 2005 Feb;25(1):93-104. doi: 10.1055/s-2005-864785. Semin Liver Dis. 2005. PMID: 15732001 Review.
-
Role of lipid metabolism in hepatitis C virus assembly and entry.Biol Cell. 2009 Oct 28;102(1):63-74. doi: 10.1042/BC20090125. Biol Cell. 2009. PMID: 19857204 Review.
Cited by
-
Characterization of hepatitis C virus particle subpopulations reveals multiple usage of the scavenger receptor BI for entry steps.J Biol Chem. 2012 Sep 7;287(37):31242-57. doi: 10.1074/jbc.M112.365924. Epub 2012 Jul 5. J Biol Chem. 2012. PMID: 22767607 Free PMC article.
-
Hepatitis C virus: virology and life cycle.Clin Mol Hepatol. 2013 Mar;19(1):17-25. doi: 10.3350/cmh.2013.19.1.17. Epub 2013 Mar 25. Clin Mol Hepatol. 2013. PMID: 23593605 Free PMC article. Review.
-
Associations between serum lipids and hepatitis C antiviral treatment efficacy.Hepatology. 2010 Sep;52(3):854-63. doi: 10.1002/hep.23796. Hepatology. 2010. PMID: 20690192 Free PMC article.
-
HCV Pit Stop at the Lipid Droplet: Refuel Lipids and Put on a Lipoprotein Coat before Exit.Cells. 2019 Mar 12;8(3):233. doi: 10.3390/cells8030233. Cells. 2019. PMID: 30871009 Free PMC article. Review.
-
Role for ADP ribosylation factor 1 in the regulation of hepatitis C virus replication.J Virol. 2011 Jan;85(2):946-56. doi: 10.1128/JVI.00753-10. Epub 2010 Nov 10. J Virol. 2011. PMID: 21068255 Free PMC article.
References
-
- Adeli, K. 1994. Regulated intracellular degradation of apolipoprotein B in semipermeable HepG2 cells. J. Biol. Chem. 2699166-9175. - PubMed
-
- Andre, P., G. Perlemuter, A. Budkowska, C. Brechot, and V. Lotteau. 2005. Hepatitis C virus particles and lipoprotein metabolism. Semin. Liver Dis. 2593-104. - PubMed
-
- Bartosch, B., A. Vitelli, C. Granier, C. Goujon, J. Dubuisson, S. Pascale, E. Scarselli, R. Cortese, A. Nicosia, and F. L. Cosset. 2003. Cell entry of hepatitis C virus requires a set of co-receptors that include the CD81 tetraspanin and the SR-B1 scavenger receptor. J. Biol. Chem. 27841624-41630. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
