

Evolution, biogenesis, expression, and target predictions of a substantially expanded set of Drosophila microRNAs
- PMID: 17989254
- PMCID: PMC2099593
- DOI: 10.1101/gr.6597907
Evolution, biogenesis, expression, and target predictions of a substantially expanded set of Drosophila microRNAs
Abstract
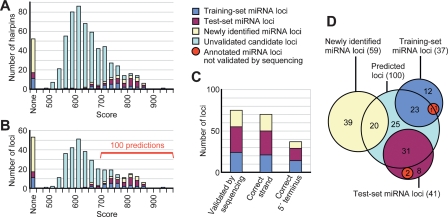
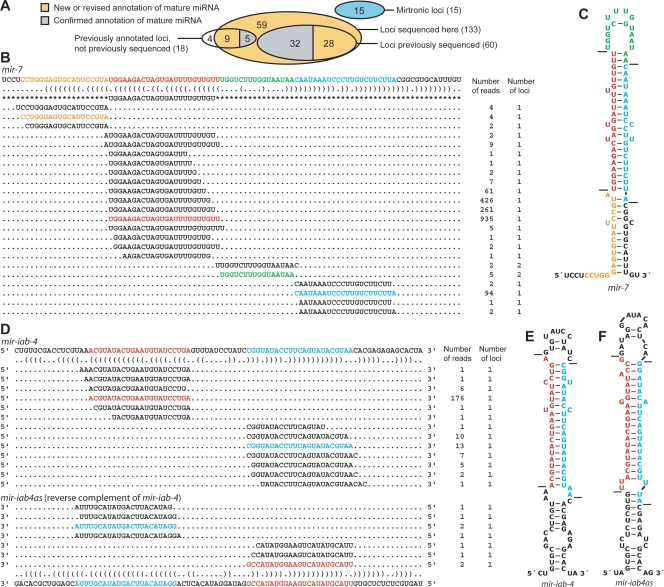
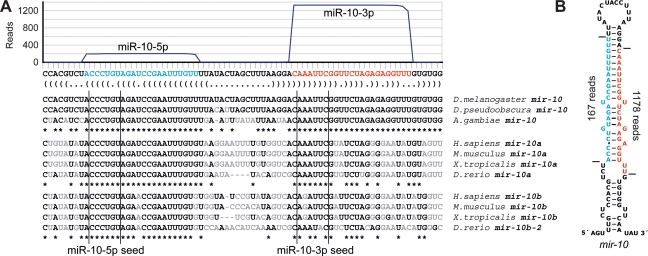
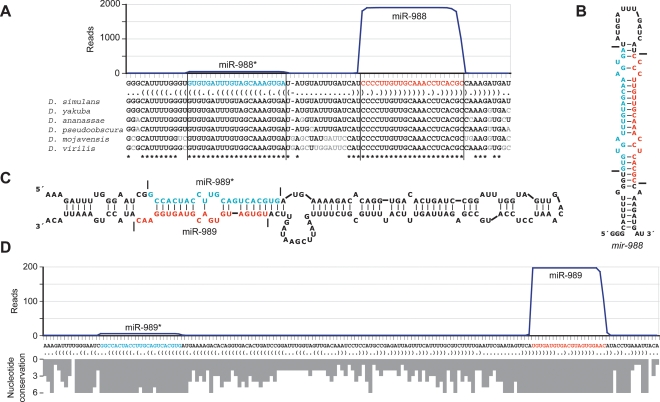
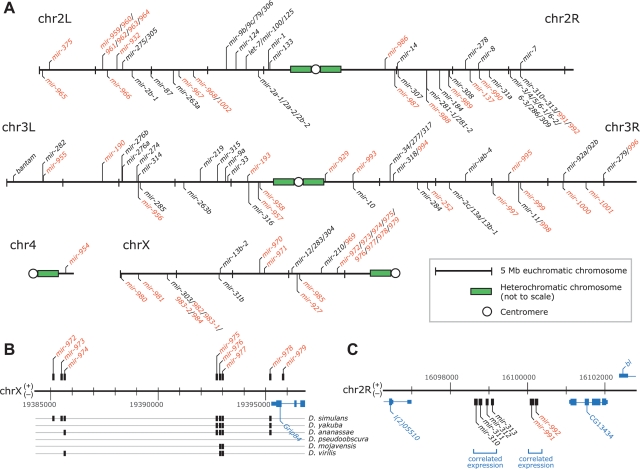
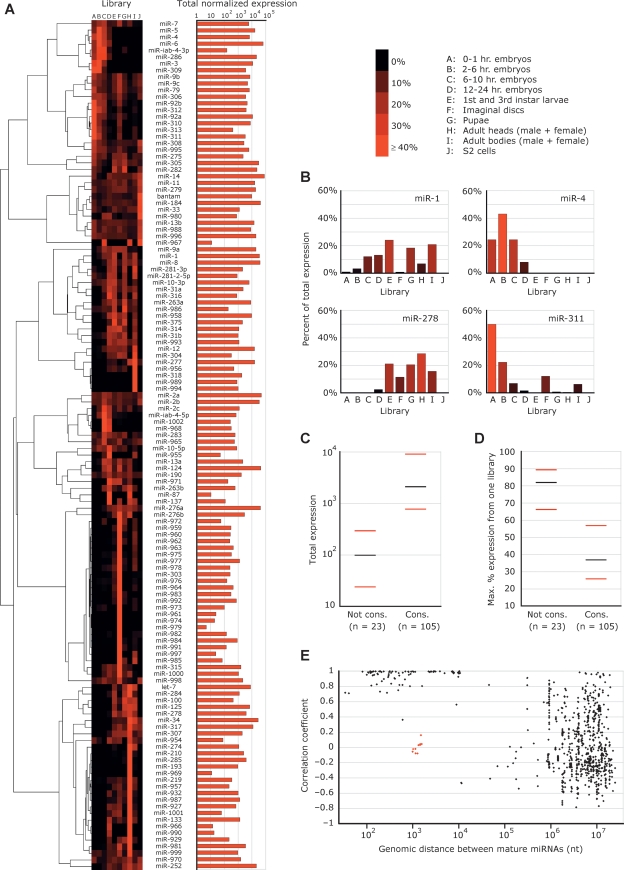
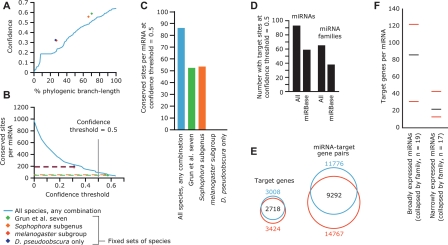
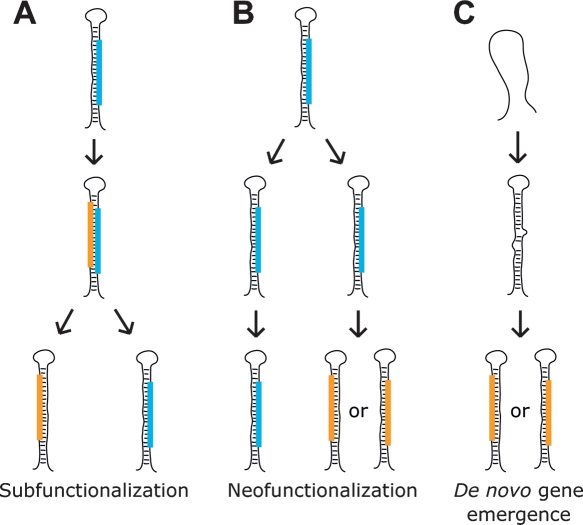
MicroRNA (miRNA) genes give rise to small regulatory RNAs in a wide variety of organisms. We used computational methods to predict miRNAs conserved among Drosophila species and large-scale sequencing of small RNAs from Drosophila melanogaster to experimentally confirm and complement these predictions. In addition to validating 20 of our top 45 predictions for novel miRNA loci, the large-scale sequencing identified many miRNAs that had not been predicted. In total, 59 novel genes were identified, increasing our tally of confirmed fly miRNAs to 148. The large-scale sequencing also refined the identities of previously known miRNAs and provided insights into their biogenesis and expression. Many miRNAs were expressed in particular developmental contexts, with a large cohort of miRNAs expressed primarily in imaginal discs. Conserved miRNAs typically were expressed more broadly and robustly than were nonconserved miRNAs, and those conserved miRNAs with more restricted expression tended to have fewer predicted targets than those expressed more broadly. Predicted targets for the expanded set of microRNAs substantially increased and revised the miRNA-target relationships that appear conserved among the fly species. Insights were also provided into miRNA gene evolution, including evidence for emergent regulatory function deriving from the opposite arm of the miRNA hairpin, exemplified by mir-10, and even the opposite strand of the DNA, exemplified by mir-iab-4.
Figures
Similar articles
-
Computational identification of Drosophila microRNA genes.Genome Biol. 2003;4(7):R42. doi: 10.1186/gb-2003-4-7-r42. Epub 2003 Jun 30. Genome Biol. 2003. PMID: 12844358 Free PMC article.
-
Identification of Drosophila MicroRNA targets.PLoS Biol. 2003 Dec;1(3):E60. doi: 10.1371/journal.pbio.0000060. Epub 2003 Oct 13. PLoS Biol. 2003. PMID: 14691535 Free PMC article.
-
Identification of novel Drosophila melanogaster microRNAs.PLoS One. 2007 Nov 28;2(11):e1265. doi: 10.1371/journal.pone.0001265. PLoS One. 2007. PMID: 18043761 Free PMC article.
-
microRNAs in Drosophila regulate cell fate by repressing single mRNA targets.Int J Dev Biol. 2017;61(3-4-5):165-170. doi: 10.1387/ijdb.160271ht. Int J Dev Biol. 2017. PMID: 28621414 Review.
-
The evolution and functional diversification of animal microRNA genes.Cell Res. 2008 Oct;18(10):985-96. doi: 10.1038/cr.2008.278. Cell Res. 2008. PMID: 18711447 Free PMC article. Review.
Cited by
-
miRNA genes of an invasive vector mosquito, Aedes albopictus.PLoS One. 2013 Jul 1;8(7):e67638. doi: 10.1371/journal.pone.0067638. Print 2013. PLoS One. 2013. PMID: 23840875 Free PMC article.
-
Prediction of conserved precursors of miRNAs and their mature forms by integrating position-specific structural features.PLoS One. 2012;7(9):e44314. doi: 10.1371/journal.pone.0044314. Epub 2012 Sep 5. PLoS One. 2012. PMID: 22957063 Free PMC article.
-
MicroRNA Transcriptomes Reveal Prevalence of Rare and Species-Specific Arm Switching Events During Zebrafish Ontogenesis.Evol Bioinform Online. 2024 Jul 24;20:11769343241263230. doi: 10.1177/11769343241263230. eCollection 2024. Evol Bioinform Online. 2024. PMID: 39055772 Free PMC article.
-
MicroRNA-276a functions in ellipsoid body and mushroom body neurons for naive and conditioned olfactory avoidance in Drosophila.J Neurosci. 2013 Mar 27;33(13):5821-33. doi: 10.1523/JNEUROSCI.4004-12.2013. J Neurosci. 2013. PMID: 23536094 Free PMC article.
-
The exonuclease Nibbler regulates age-associated traits and modulates piRNA length in Drosophila.Aging Cell. 2015 Jun;14(3):443-52. doi: 10.1111/acel.12323. Epub 2015 Mar 6. Aging Cell. 2015. PMID: 25754031 Free PMC article.
References
-
- Abbott A.L., Alvarez-Saavedra E., Miska E.A., Lau N.C., Bartel D.P., Horvitz H.R., Ambros V., Alvarez-Saavedra E., Miska E.A., Lau N.C., Bartel D.P., Horvitz H.R., Ambros V., Miska E.A., Lau N.C., Bartel D.P., Horvitz H.R., Ambros V., Lau N.C., Bartel D.P., Horvitz H.R., Ambros V., Bartel D.P., Horvitz H.R., Ambros V., Horvitz H.R., Ambros V., Ambros V. The let-7 microRNA family members mir-48, mir-84, and mir-241 function together to regulate developmental timing in Caenorhabditis elegans. Dev. Cell. 2005;9:403–414. - PMC - PubMed
-
- Adams M.D., Celniker S.E., Holt R.A., Evans C.A., Gocayne J.D., Amanatides P.G., Scherer S.E., Li P.W., Hoskins R.A., Galle R.F., Celniker S.E., Holt R.A., Evans C.A., Gocayne J.D., Amanatides P.G., Scherer S.E., Li P.W., Hoskins R.A., Galle R.F., Holt R.A., Evans C.A., Gocayne J.D., Amanatides P.G., Scherer S.E., Li P.W., Hoskins R.A., Galle R.F., Evans C.A., Gocayne J.D., Amanatides P.G., Scherer S.E., Li P.W., Hoskins R.A., Galle R.F., Gocayne J.D., Amanatides P.G., Scherer S.E., Li P.W., Hoskins R.A., Galle R.F., Amanatides P.G., Scherer S.E., Li P.W., Hoskins R.A., Galle R.F., Scherer S.E., Li P.W., Hoskins R.A., Galle R.F., Li P.W., Hoskins R.A., Galle R.F., Hoskins R.A., Galle R.F., Galle R.F., et al. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–2195. - PubMed
-
- Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J., Gish W., Miller W., Myers E.W., Lipman D.J., Miller W., Myers E.W., Lipman D.J., Myers E.W., Lipman D.J., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. - PubMed
-
- Ambros V., Lee R.C., Lavanway A., Williams P.T., Jewell D., Lee R.C., Lavanway A., Williams P.T., Jewell D., Lavanway A., Williams P.T., Jewell D., Williams P.T., Jewell D., Jewell D. MicroRNAs and other tiny endogenous RNAs in C. elegans. Curr. Biol. 2003;13:807–818. - PubMed
-
- Aravin A.A., Lagos-Quintana M., Yalcin A., Zavolan M., Marks D., Snyder B., Gaasterland T., Meyer J., Tuschl T., Lagos-Quintana M., Yalcin A., Zavolan M., Marks D., Snyder B., Gaasterland T., Meyer J., Tuschl T., Yalcin A., Zavolan M., Marks D., Snyder B., Gaasterland T., Meyer J., Tuschl T., Zavolan M., Marks D., Snyder B., Gaasterland T., Meyer J., Tuschl T., Marks D., Snyder B., Gaasterland T., Meyer J., Tuschl T., Snyder B., Gaasterland T., Meyer J., Tuschl T., Gaasterland T., Meyer J., Tuschl T., Meyer J., Tuschl T., Tuschl T. The small RNA profile during Drosophila melanogaster development. Dev. Cell. 2003;5:337–350. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases