

28-way vertebrate alignment and conservation track in the UCSC Genome Browser
- PMID: 17984227
- PMCID: PMC2099589
- DOI: 10.1101/gr.6761107
28-way vertebrate alignment and conservation track in the UCSC Genome Browser
Abstract
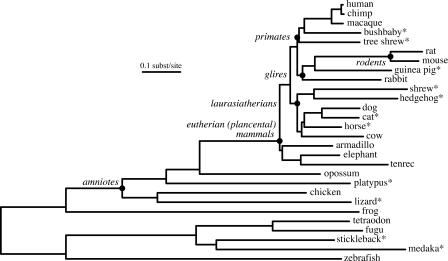
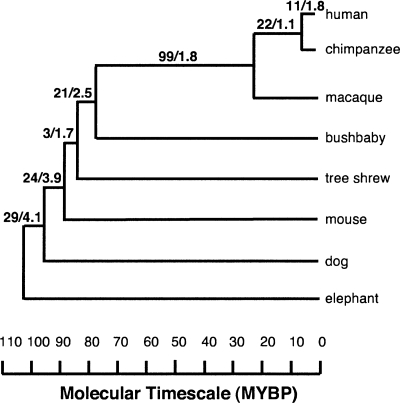
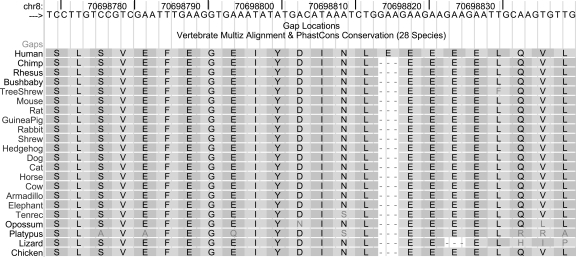
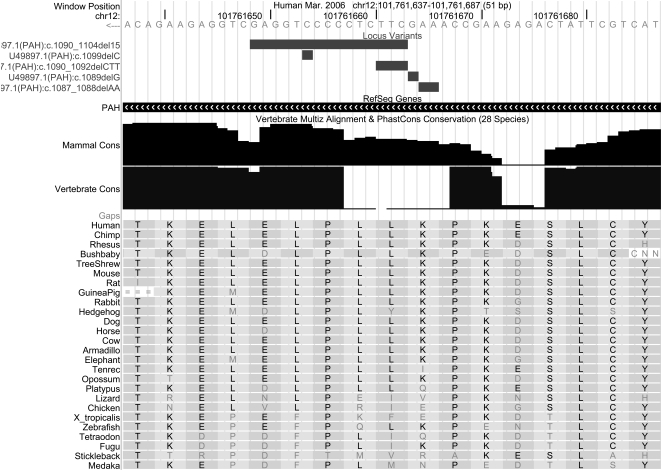
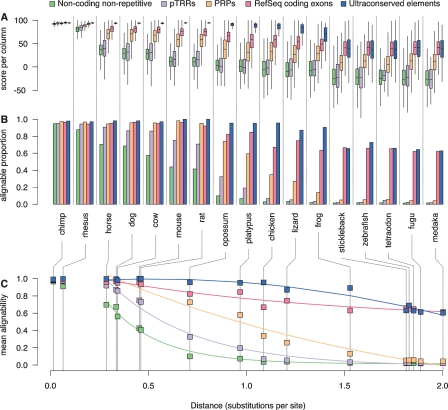
This article describes a set of alignments of 28 vertebrate genome sequences that is provided by the UCSC Genome Browser. The alignments can be viewed on the Human Genome Browser (March 2006 assembly) at http://genome.ucsc.edu, downloaded in bulk by anonymous FTP from http://hgdownload.cse.ucsc.edu/goldenPath/hg18/multiz28way, or analyzed with the Galaxy server at http://g2.bx.psu.edu. This article illustrates the power of this resource for exploring vertebrate and mammalian evolution, using three examples. First, we present several vignettes involving insertions and deletions within protein-coding regions, including a look at some human-specific indels. Then we study the extent to which start codons and stop codons in the human sequence are conserved in other species, showing that start codons are in general more poorly conserved than stop codons. Finally, an investigation of the phylogenetic depth of conservation for several classes of functional elements in the human genome reveals striking differences in the rates and modes of decay in alignability. Each functional class has a distinctive period of stringent constraint, followed by decays that allow (for the case of regulatory regions) or reject (for coding regions and ultraconserved elements) insertions and deletions.
Figures


Similar articles
-
Computational prediction of cis-regulatory modules from multispecies alignments using Galaxy, Table Browser, and GALA.Methods Mol Biol. 2006;338:91-103. doi: 10.1385/1-59745-097-9:91. Methods Mol Biol. 2006. PMID: 16888352
-
Track data hubs enable visualization of user-defined genome-wide annotations on the UCSC Genome Browser.Bioinformatics. 2014 Apr 1;30(7):1003-5. doi: 10.1093/bioinformatics/btt637. Epub 2013 Nov 13. Bioinformatics. 2014. PMID: 24227676 Free PMC article.
-
Mulan: multiple-sequence local alignment and visualization for studying function and evolution.Genome Res. 2005 Jan;15(1):184-94. doi: 10.1101/gr.3007205. Epub 2004 Dec 8. Genome Res. 2005. PMID: 15590941 Free PMC article.
-
The UCSC genome browser database: update 2007.Nucleic Acids Res. 2007 Jan;35(Database issue):D668-73. doi: 10.1093/nar/gkl928. Epub 2006 Nov 16. Nucleic Acids Res. 2007. PMID: 17142222 Free PMC article.
-
The UCSC Genome Browser database: 2014 update.Nucleic Acids Res. 2014 Jan;42(Database issue):D764-70. doi: 10.1093/nar/gkt1168. Epub 2013 Nov 21. Nucleic Acids Res. 2014. PMID: 24270787 Free PMC article.
Cited by
-
Activation of an endogenous retrovirus-associated long non-coding RNA in human adenocarcinoma.Genome Med. 2015 Mar 5;7(1):22. doi: 10.1186/s13073-015-0142-6. eCollection 2015. Genome Med. 2015. PMID: 25821520 Free PMC article.
-
Sequence shortening in the rodent ancestor.Genome Res. 2012 Mar;22(3):478-85. doi: 10.1101/gr.121897.111. Epub 2011 Nov 29. Genome Res. 2012. PMID: 22128134 Free PMC article.
-
Simultaneous SNP identification and assessment of allele-specific bias from ChIP-seq data.BMC Genet. 2012 Sep 5;13:46. doi: 10.1186/1471-2156-13-46. BMC Genet. 2012. PMID: 22950704 Free PMC article.
-
Regulation of ALF promoter activity in Xenopus oocytes.PLoS One. 2009 Aug 17;4(8):e6664. doi: 10.1371/journal.pone.0006664. PLoS One. 2009. PMID: 19684851 Free PMC article.
-
A map of open chromatin in human pancreatic islets.Nat Genet. 2010 Mar;42(3):255-9. doi: 10.1038/ng.530. Epub 2010 Jan 31. Nat Genet. 2010. PMID: 20118932 Free PMC article.
References
-
- Bejerano G., Pheasant M., Makunin I., Stephen S., Kent W.J., Mattick J.S., Haussler D., Pheasant M., Makunin I., Stephen S., Kent W.J., Mattick J.S., Haussler D., Makunin I., Stephen S., Kent W.J., Mattick J.S., Haussler D., Stephen S., Kent W.J., Mattick J.S., Haussler D., Kent W.J., Mattick J.S., Haussler D., Mattick J.S., Haussler D., Haussler D. Ultraconserved elements in the human genome. Science. 2004;304:1321–1325. - PubMed
-
- Blanchette M., Kent W.J., Riemer C., Elnitski L., Smit A.F.A., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D., Kent W.J., Riemer C., Elnitski L., Smit A.F.A., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D., Riemer C., Elnitski L., Smit A.F.A., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D., Elnitski L., Smit A.F.A., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D., Smit A.F.A., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D., Baertsch R., Rosenbloom K., Clawson H., Green E.D., Rosenbloom K., Clawson H., Green E.D., Clawson H., Green E.D., Green E.D., et al. Aligning multiple genomic sequences with the Threaded Blockset Aligner. Genome Res. 2004;14:708–715. - PMC - PubMed
-
- Blanchette M., Bataille A.R., Chen X., Poitras C., Laganiere J., Lefebvre C., Deblois G., Giguere V., Ferretti V., Bergeron D., Bataille A.R., Chen X., Poitras C., Laganiere J., Lefebvre C., Deblois G., Giguere V., Ferretti V., Bergeron D., Chen X., Poitras C., Laganiere J., Lefebvre C., Deblois G., Giguere V., Ferretti V., Bergeron D., Poitras C., Laganiere J., Lefebvre C., Deblois G., Giguere V., Ferretti V., Bergeron D., Laganiere J., Lefebvre C., Deblois G., Giguere V., Ferretti V., Bergeron D., Lefebvre C., Deblois G., Giguere V., Ferretti V., Bergeron D., Deblois G., Giguere V., Ferretti V., Bergeron D., Giguere V., Ferretti V., Bergeron D., Ferretti V., Bergeron D., Bergeron D., et al. Genome-wide computational prediction of transcriptional regulatory modules reveals new insights into human gene expression. Genome Res. 2006;16:656–668. - PMC - PubMed
-
- Blankenberg D., Taylor J., Schenck I., He J., Zhang Y., Ghent M., Veeraaghavan N., Albert I., Miller W., Makova K., Taylor J., Schenck I., He J., Zhang Y., Ghent M., Veeraaghavan N., Albert I., Miller W., Makova K., Schenck I., He J., Zhang Y., Ghent M., Veeraaghavan N., Albert I., Miller W., Makova K., He J., Zhang Y., Ghent M., Veeraaghavan N., Albert I., Miller W., Makova K., Zhang Y., Ghent M., Veeraaghavan N., Albert I., Miller W., Makova K., Ghent M., Veeraaghavan N., Albert I., Miller W., Makova K., Veeraaghavan N., Albert I., Miller W., Makova K., Albert I., Miller W., Makova K., Miller W., Makova K., Makova K., et al. A framework for collaborative analysis of ENCODE data: Making large-scale analyses biologist-friendly. Genome Res. 2007;17:960–964. - PMC - PubMed
-
- Diallo A.B., Makarenkov V., Blanchette M., Makarenkov V., Blanchette M., Blanchette M. Exact and heuristic algorithms for the Indel Maximum Likelihood problem. J. Comput. Biol. 2007;14:446–461. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources