

Reactivation of Kaposi's sarcoma-associated herpesvirus from latency requires MEK/ERK, JNK and p38 multiple mitogen-activated protein kinase pathways
- PMID: 17964626
- PMCID: PMC2239004
- DOI: 10.1016/j.virol.2007.09.040
Reactivation of Kaposi's sarcoma-associated herpesvirus from latency requires MEK/ERK, JNK and p38 multiple mitogen-activated protein kinase pathways
Abstract
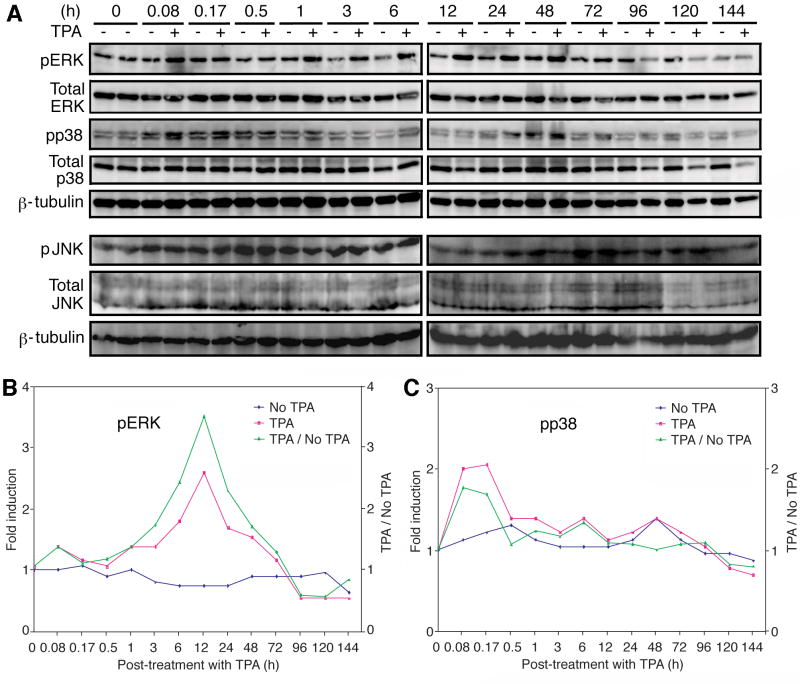
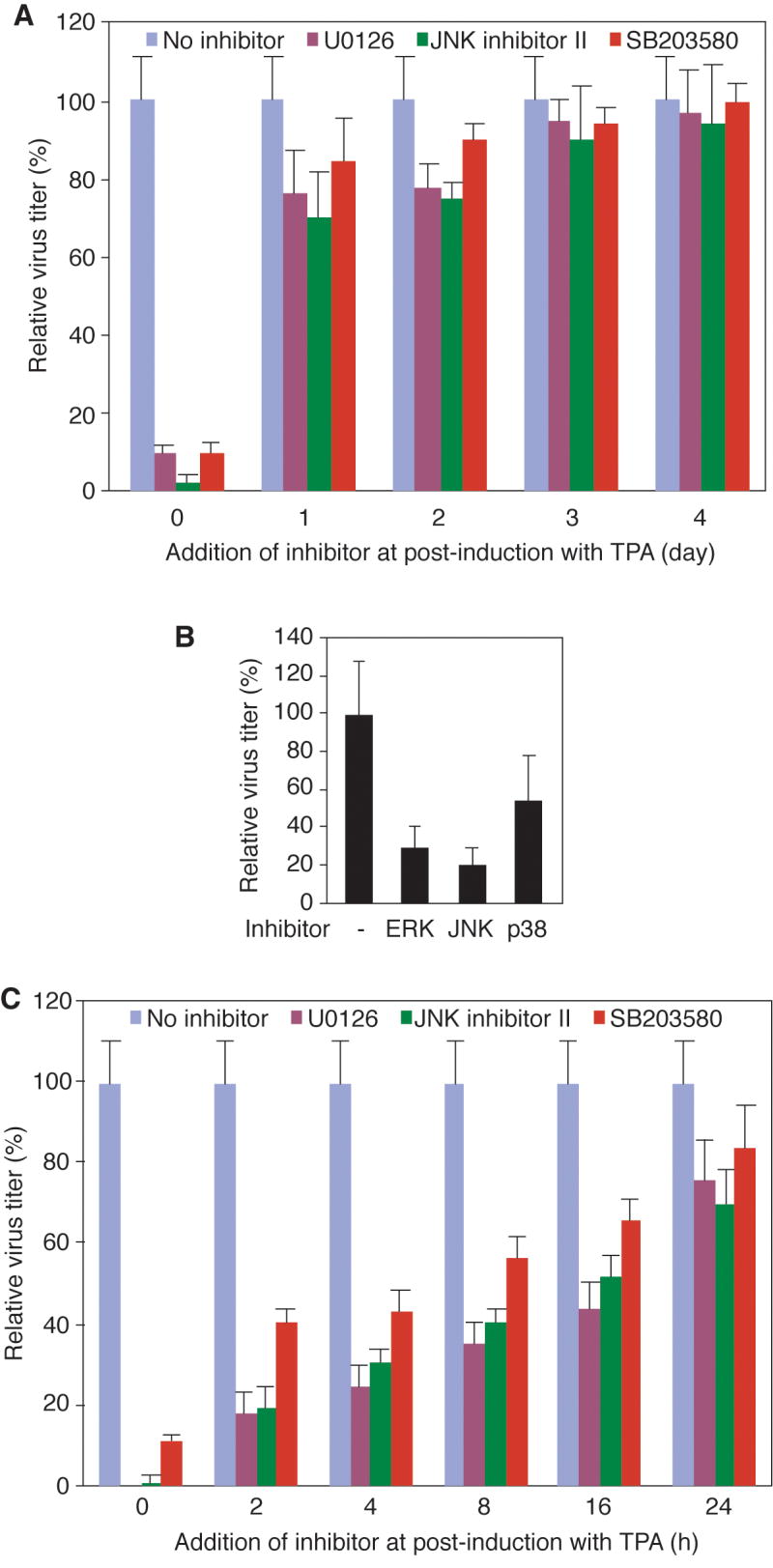
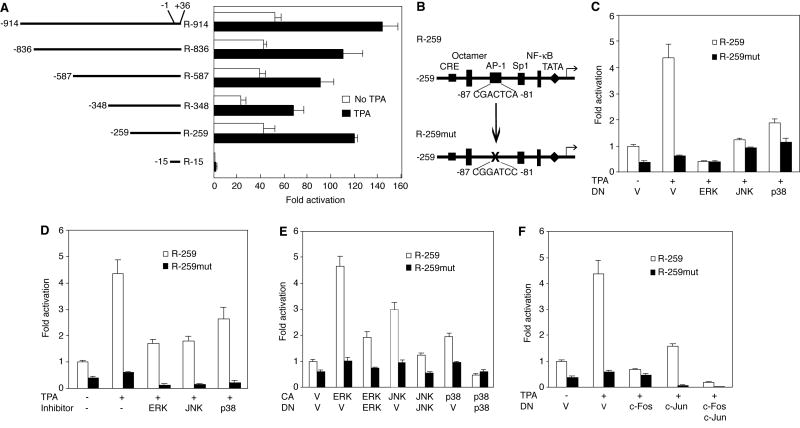
Lytic replication of Kaposi's sarcoma-associated herpesvirus (KSHV) promotes the progression of Kaposi's sarcoma (KS), a dominant malignancy in patients with AIDS. While 12-O-tetradecanoyl-phorbol-13-acetate (TPA)-induced KSHV reactivation from latency is mediated by the protein kinase C delta and MEK/ERK mitogen-activated protein kinase (MAPK) pathways, we have recently shown that the MEK/ERK, JNK and p38 MAPK pathways modulate KSHV lytic replication during productive primary infection of human umbilical vein endothelial cells [Pan, H., Xie, J., Ye, F., Gao, S.J., 2006. Modulation of Kaposi's sarcoma-associated herpesvirus infection and replication by MEK/ERK, JNK, and p38 multiple mitogen-activated protein kinase pathways during primary infection. J. Virol. 80 (11), 5371-5382]. Here, we report that, besides the MEK/ERK pathway, the JNK and p38 MAPK pathways also mediate TPA-induced KSHV reactivation from latency. The MEK/ERK, JNK and p38 MAPK pathways were constitutively activated in latent KSHV-infected BCBL-1 cells. TPA treatment enhanced the levels of activated ERK and p38 but not those of activated JNK. Inhibitors of all three MAPK pathways reduced TPA-induced production of KSHV infectious virions in BCBL-1 cells in a dose-dependent fashion. The inhibitors blocked KSHV lytic replication at the early stage(s) of reactivation, and reduced the expression of viral lytic genes including RTA, a key immediate-early transactivator of viral lytic replication. Activation of MAPK pathways was necessary and sufficient for activating the promoter of RTA. Furthermore, we showed that the activation of RTA promoter by MAPK pathways was mediated by their downstream target AP-1. Together, these findings suggest that MAPK pathways might have general roles in regulating the life cycle of KSHV by mediating both viral infection and switch from viral latency to lytic replication.
Figures




Similar articles
-
Activation of Kaposi's sarcoma-associated herpesvirus (KSHV) by inhibitors of class III histone deacetylases: identification of sirtuin 1 as a regulator of the KSHV life cycle.J Virol. 2014 Jun;88(11):6355-67. doi: 10.1128/JVI.00219-14. Epub 2014 Mar 26. J Virol. 2014. PMID: 24672028 Free PMC article.
-
Modulation of Kaposi's sarcoma-associated herpesvirus infection and replication by MEK/ERK, JNK, and p38 multiple mitogen-activated protein kinase pathways during primary infection.J Virol. 2006 Jun;80(11):5371-82. doi: 10.1128/JVI.02299-05. J Virol. 2006. PMID: 16699017 Free PMC article.
-
ERK1/2 and MEK1/2 induced by Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) early during infection of target cells are essential for expression of viral genes and for establishment of infection.J Virol. 2005 Aug;79(16):10308-29. doi: 10.1128/JVI.79.16.10308-10329.2005. J Virol. 2005. PMID: 16051824 Free PMC article.
-
Reactivation and Lytic Replication of Kaposi's Sarcoma-Associated Herpesvirus: An Update.Front Microbiol. 2017 Apr 20;8:613. doi: 10.3389/fmicb.2017.00613. eCollection 2017. Front Microbiol. 2017. PMID: 28473805 Free PMC article. Review.
-
Regulation of KSHV Latency and Lytic Reactivation.Viruses. 2020 Sep 17;12(9):1034. doi: 10.3390/v12091034. Viruses. 2020. PMID: 32957532 Free PMC article. Review.
Cited by
-
Kinome profiling of non-Hodgkin lymphoma identifies Tyro3 as a therapeutic target in primary effusion lymphoma.Proc Natl Acad Sci U S A. 2019 Aug 13;116(33):16541-16550. doi: 10.1073/pnas.1903991116. Epub 2019 Jul 25. Proc Natl Acad Sci U S A. 2019. PMID: 31346082 Free PMC article.
-
Molecular biology of Kaposi's sarcoma-associated herpesvirus and related oncogenesis.Adv Virus Res. 2010;78:87-142. doi: 10.1016/B978-0-12-385032-4.00003-3. Adv Virus Res. 2010. PMID: 21040832 Free PMC article. Review.
-
Activation of Kaposi's sarcoma-associated herpesvirus (KSHV) by inhibitors of class III histone deacetylases: identification of sirtuin 1 as a regulator of the KSHV life cycle.J Virol. 2014 Jun;88(11):6355-67. doi: 10.1128/JVI.00219-14. Epub 2014 Mar 26. J Virol. 2014. PMID: 24672028 Free PMC article.
-
Screening of the Human Kinome Identifies MSK1/2-CREB1 as an Essential Pathway Mediating Kaposi's Sarcoma-Associated Herpesvirus Lytic Replication during Primary Infection.J Virol. 2015 Sep;89(18):9262-80. doi: 10.1128/JVI.01098-15. Epub 2015 Jun 24. J Virol. 2015. PMID: 26109721 Free PMC article.
-
Amplification of JNK signaling is necessary to complete the murine gammaherpesvirus 68 lytic replication cycle.J Virol. 2012 Dec;86(24):13253-62. doi: 10.1128/JVI.01432-12. Epub 2012 Sep 26. J Virol. 2012. PMID: 23015701 Free PMC article.
References
-
- Akula SM, Pramod NP, Wang FZ, Chandran B. Integrin alpha3beta1 (CD 49c/29) is a cellular receptor for Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) entry into the target cells. Cell. 2002;108(3):407–419. - PubMed
-
- An J, Lichtenstein A, G B, Rettig M. The Kaposi's sarcoma-associated herpesvirus (KSHV) induces cellular interleukin 6 expression: role of the KSHV latency-associated nuclear antigen and the AP1 response element. Blood. 2002;99(2):649–654. - PubMed
-
- An J, Sun Y, Rettig MB. Transcriptional coactivation of c-Jun by the KSHV-encoded LANA. Blood. 2004;103(1):222–228. - PubMed
-
- An J, Sun Y, Sun R, Rettig MB. Kaposi's sarcoma-associated herpesvirus encoded vFLIP induces cellular IL-6 expression: the role of the NF-kappaB and JNK/AP1 pathways. Oncogene. 2003;22(22):3371–3385. - PubMed
-
- Aoki H, Ohnishi H, Hama K, Ishijima T, Satoh Y, Hanatsuka K, Ohashi A, Wada S, Miyata T, Kita H, Yamamoto H, Osawa H, Sato K, Tamada K, Yasuda H, Mashima H, Sugano K. Autocrine loop between TGF-beta1 and IL-1beta through Smad3- and ERK-dependent pathways in rat pancreatic stellate cells. Am J Physiol Cell Physiol. 2006;290(4):C1100–1108. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 CA096512/CA/NCI NIH HHS/United States
- R01 DE017333-02/DE/NIDCR NIH HHS/United States
- R01 CA124332/CA/NCI NIH HHS/United States
- R01 CA096512-05/CA/NCI NIH HHS/United States
- DE017333/DE/NIDCR NIH HHS/United States
- CA096512/CA/NCI NIH HHS/United States
- M01 RR001346-258134/RR/NCRR NIH HHS/United States
- R01 DE017333/DE/NIDCR NIH HHS/United States
- R01 CA124332-01A2/CA/NCI NIH HHS/United States
- R01 CA096512-04/CA/NCI NIH HHS/United States
- R01 CA132637/CA/NCI NIH HHS/United States
- R01 CA124332-02/CA/NCI NIH HHS/United States
- M01 RR001346/RR/NCRR NIH HHS/United States
- CA124332/CA/NCI NIH HHS/United States
- R01 DE017333-01/DE/NIDCR NIH HHS/United States
- R01 DE017333-03/DE/NIDCR NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
