

Dysfunctional telomeres activate an ATM-ATR-dependent DNA damage response to suppress tumorigenesis
- PMID: 17948054
- PMCID: PMC2080807
- DOI: 10.1038/sj.emboj.7601893
Dysfunctional telomeres activate an ATM-ATR-dependent DNA damage response to suppress tumorigenesis
Abstract
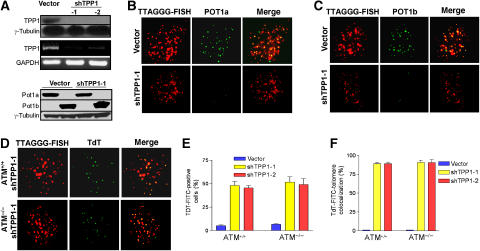
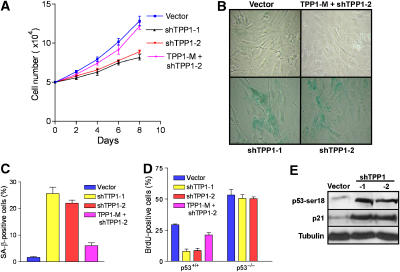
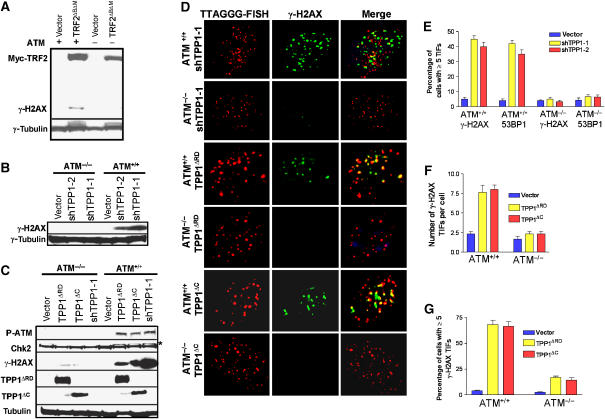
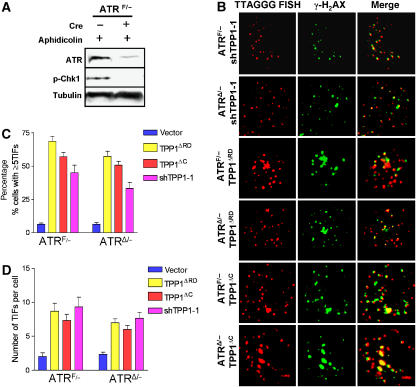
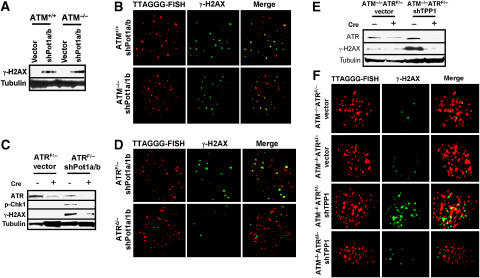
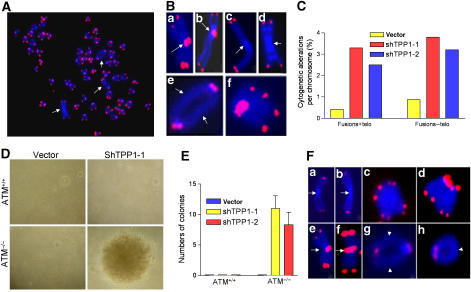
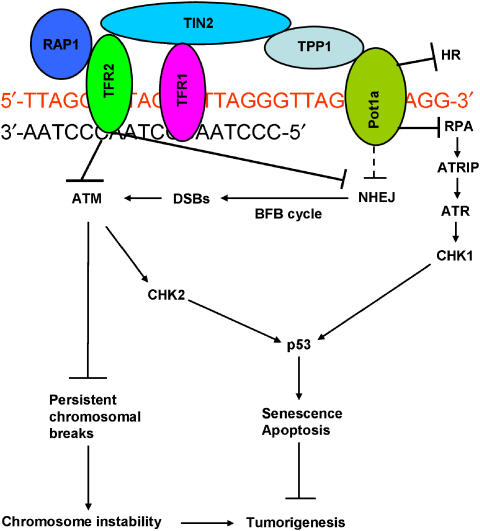
The POT1 (protection of telomeres) protein binds the single-stranded G-rich overhang and is essential for both telomere end protection and telomere length regulation. Telomeric binding of POT1 is enhanced by its interaction with TPP1. In this study, we demonstrate that mouse Tpp1 confers telomere end protection by recruiting Pot1a and Pot1b to telomeres. Knockdown of Tpp1 elicits a p53-dependent growth arrest and an ATM-dependent DNA damage response at telomeres. In contrast to depletion of Trf2, which activates ATM, removal of Pot1a and Pot1b from telomeres initiates an ATR-dependent DNA damage response (DDR). Finally, we show that telomere dysfunction as a result of Tpp1 depletion promotes chromosomal instability and tumorigenesis in the absence of an ATM-dependent DDR. Our results uncover a novel ATR-dependent DDR at telomeres that is normally shielded by POT1 binding to the single-stranded G-overhang. In addition, our results suggest that loss of ATM can cooperate with dysfunctional telomeres to promote cellular transformation and tumor formation in vivo.
Figures
Similar articles
-
Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1.Nature. 2007 Aug 30;448(7157):1068-71. doi: 10.1038/nature06065. Epub 2007 Aug 8. Nature. 2007. PMID: 17687332
-
Multiple roles for MRE11 at uncapped telomeres.Nature. 2009 Aug 13;460(7257):914-8. doi: 10.1038/nature08196. Epub 2009 Jul 26. Nature. 2009. PMID: 19633651 Free PMC article.
-
Telomere protection by TPP1/POT1 requires tethering to TIN2.Mol Cell. 2011 Nov 18;44(4):647-59. doi: 10.1016/j.molcel.2011.08.043. Mol Cell. 2011. PMID: 22099311 Free PMC article.
-
The Connection Between Cell Fate and Telomere.Adv Exp Med Biol. 2021;1275:71-100. doi: 10.1007/978-3-030-49844-3_3. Adv Exp Med Biol. 2021. PMID: 33539012 Review.
-
Structural biology of telomeres and telomerase.Cell Mol Life Sci. 2020 Jan;77(1):61-79. doi: 10.1007/s00018-019-03369-x. Epub 2019 Nov 14. Cell Mol Life Sci. 2020. PMID: 31728577 Free PMC article. Review.
Cited by
-
Telomere- and Telomerase-Associated Proteins and Their Functions in the Plant Cell.Front Plant Sci. 2016 Jun 28;7:851. doi: 10.3389/fpls.2016.00851. eCollection 2016. Front Plant Sci. 2016. PMID: 27446102 Free PMC article. Review.
-
Five dysfunctional telomeres predict onset of senescence in human cells.EMBO Rep. 2011 Dec 23;13(1):52-9. doi: 10.1038/embor.2011.227. EMBO Rep. 2011. PMID: 22157895 Free PMC article.
-
Conserved telomere maintenance component 1 interacts with STN1 and maintains chromosome ends in higher eukaryotes.Mol Cell. 2009 Oct 23;36(2):207-18. doi: 10.1016/j.molcel.2009.09.017. Mol Cell. 2009. PMID: 19854131 Free PMC article.
-
The protein network surrounding the human telomere repeat binding factors TRF1, TRF2, and POT1.PLoS One. 2010 Aug 25;5(8):e12407. doi: 10.1371/journal.pone.0012407. PLoS One. 2010. PMID: 20811636 Free PMC article.
-
Short telomeres are sufficient to cause the degenerative defects associated with aging.Am J Hum Genet. 2009 Dec;85(6):823-32. doi: 10.1016/j.ajhg.2009.10.028. Am J Hum Genet. 2009. PMID: 19944403 Free PMC article.
References
-
- Bartek J, Lukas J (2003) Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 3: 421–429 - PubMed
-
- Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, Orntoft T, Lukas J, Bartek J (2005) DNA damage response as a candidate anticancer barrier in early human tumorigenesis. Nature 434: 864–870 - PubMed
-
- Bassing CH, Suh H, Ferguson DO, Chua KF, Manis J, Eckersdorff M, Gleason M, Bronson R, Lee C, Alt FW (2003) Histone H2AX: a dosage-dependent suppressor of oncogenic translocations and tumors. Cell 114: 359–370 - PubMed
-
- Brummelkamp TR, Bernards R, Agami R (2002) A system for stable expression of short interfering RNAs in mammalian cells. Science 296: 550–553 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
