

Biomechanical signals inhibit IKK activity to attenuate NF-kappaB transcription activity in inflamed chondrocytes
- PMID: 17907174
- PMCID: PMC4950916
- DOI: 10.1002/art.22933
Biomechanical signals inhibit IKK activity to attenuate NF-kappaB transcription activity in inflamed chondrocytes
Abstract
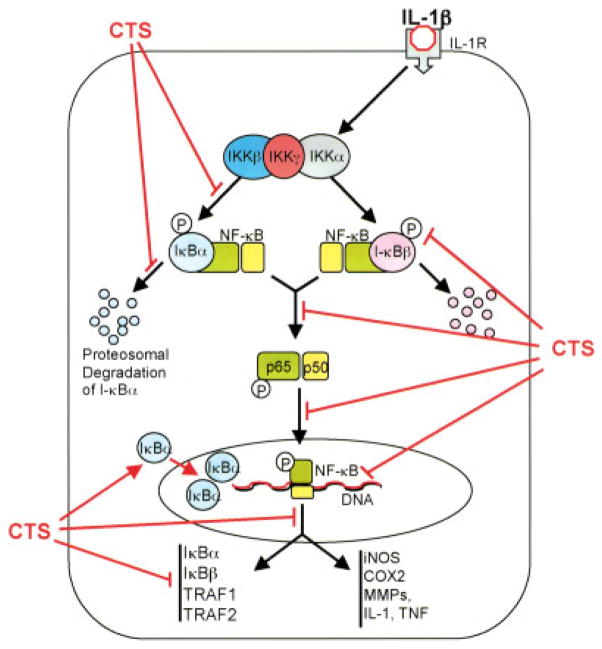
Objective: While the effects of biomechanical signals in the form of joint movement and exercise are known to be beneficial to inflamed joints, limited information is available regarding the intracellular mechanisms of their actions. This study was undertaken to examine the intracellular mechanisms by which biomechanical signals suppress proinflammatory gene induction by the interleukin-1-beta (IL-1beta)-induced NF-kappaB signaling cascade in articular chondrocytes.
Methods: Primary rat articular chondrocytes were exposed to biomechanical signals in the form of cyclic tensile strain, and the effects on the NF-kappaB signaling cascade were examined by Western blot analysis, real-time polymerase chain reaction, and immunofluorescence.
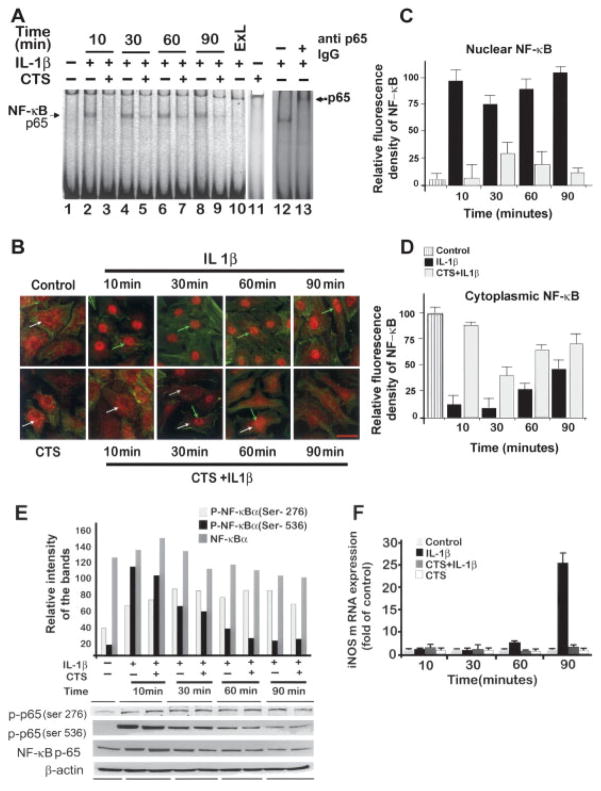
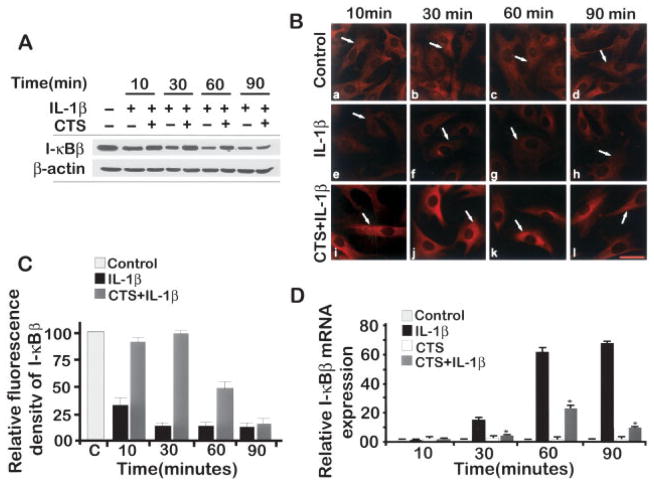
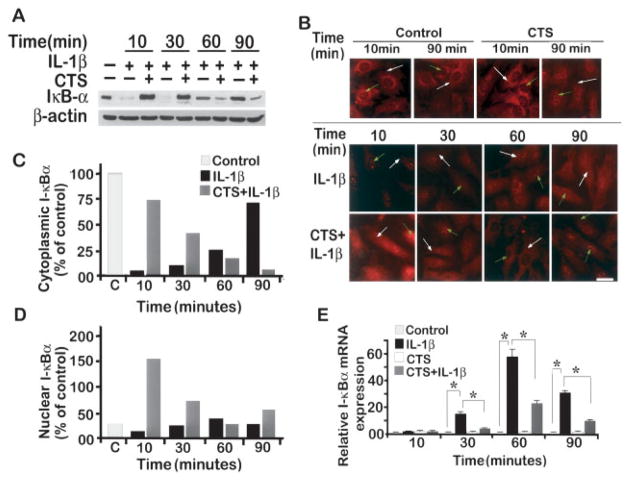
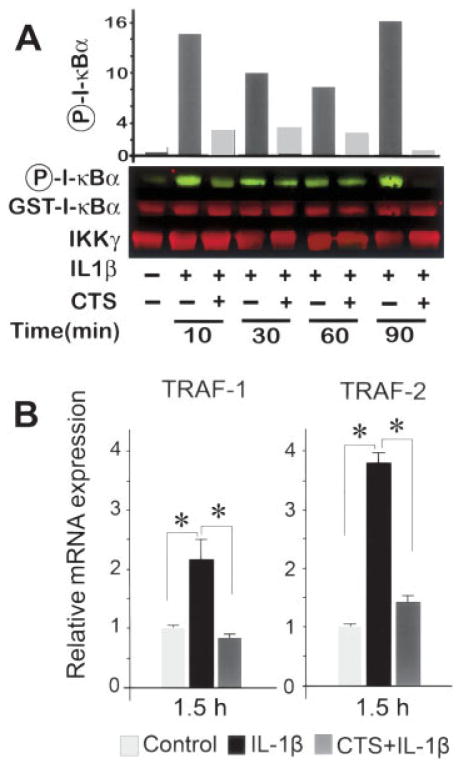
Results: Cyclic tensile strain rapidly inhibited the IL-1beta-induced nuclear translocation of NF-kappaB, but not its IL-1beta-induced phosphorylation at serine 276 and serine 536, which are necessary for its transactivation and transcriptional efficacy, respectively. Examination of upstream events revealed that cyclic tensile strain also inhibited the cytoplasmic protein degradation of IkappaBbeta and IkappaBalpha, as well as repressed their gene transcription. Additionally, cyclic tensile strain induced a rapid nuclear translocation of IkappaBalpha to potentially prevent NF-kappaB binding to DNA. Furthermore, the inhibition of IL-1beta-induced degradation of IkappaB by cyclic tensile strain was mediated by down-regulation of IkappaB kinase activity.
Conclusion: These results indicate that the signals generated by cyclic tensile strain act at multiple sites within the NF-kappaB signaling cascade to inhibit IL-1beta-induced proinflammatory gene induction. Taken together, these findings provide insight into how biomechanical signals regulate and reduce inflammation, and underscore their potential in enhancing the ability of chondrocytes to curb inflammation in diseased joints.
Figures
Comment in
-
Molecular basis of the interaction of inflammation and exercise: keep on walking!Arthritis Rheum. 2007 Oct;56(10):3176-9. doi: 10.1002/art.22934. Arthritis Rheum. 2007. PMID: 17907161 No abstract available.
Similar articles
-
Biomechanical signals suppress TAK1 activation to inhibit NF-kappaB transcriptional activation in fibrochondrocytes.J Immunol. 2007 Nov 1;179(9):6246-54. doi: 10.4049/jimmunol.179.9.6246. J Immunol. 2007. PMID: 17947700 Free PMC article.
-
Role of NF-kappaB transcription factors in antiinflammatory and proinflammatory actions of mechanical signals.Arthritis Rheum. 2004 Nov;50(11):3541-8. doi: 10.1002/art.20601. Arthritis Rheum. 2004. PMID: 15529376 Free PMC article.
-
A Polyphenol-rich Pomegranate Fruit Extract Suppresses NF-κB and IL-6 Expression by Blocking the Activation of IKKβ and NIK in Primary Human Chondrocytes.Phytother Res. 2017 May;31(5):778-782. doi: 10.1002/ptr.5799. Epub 2017 Mar 9. Phytother Res. 2017. PMID: 28276100 Free PMC article.
-
Regulation of chondrocytic gene expression by biomechanical signals.Crit Rev Eukaryot Gene Expr. 2008;18(2):139-50. doi: 10.1615/critreveukargeneexpr.v18.i2.30. Crit Rev Eukaryot Gene Expr. 2008. PMID: 18304028 Free PMC article. Review.
-
Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity.Annu Rev Immunol. 2000;18:621-63. doi: 10.1146/annurev.immunol.18.1.621. Annu Rev Immunol. 2000. PMID: 10837071 Review.
Cited by
-
Effects of cyclic tensile strain on chondrocyte metabolism: a systematic review.PLoS One. 2015 Mar 30;10(3):e0119816. doi: 10.1371/journal.pone.0119816. eCollection 2015. PLoS One. 2015. PMID: 25822615 Free PMC article. Review.
-
Mechanotransduction and cartilage integrity.Ann N Y Acad Sci. 2011 Dec;1240:32-7. doi: 10.1111/j.1749-6632.2011.06301.x. Ann N Y Acad Sci. 2011. PMID: 22172037 Free PMC article. Review.
-
Regulation of biomechanical signals by NF-kappaB transcription factors in chondrocytes.Biorheology. 2008;45(3-4):245-56. Biorheology. 2008. PMID: 18836228 Free PMC article. Review.
-
Mechanical Loading of Articular Cartilage Reduces IL-1-Induced Enzyme Expression.Cartilage. 2011 Oct 1;2(4):364-373. doi: 10.1177/1947603511407484. Cartilage. 2011. PMID: 22039566 Free PMC article.
-
Current Trends in the Evaluation of Osteochondral Lesion Treatments: Histology, Histomorphometry, and Biomechanics in Preclinical Models.Biomed Res Int. 2019 Oct 9;2019:4040236. doi: 10.1155/2019/4040236. eCollection 2019. Biomed Res Int. 2019. PMID: 31687388 Free PMC article. Review.
References
-
- Roos EM, Dahlberg L. Positive effects of moderate exercise on glycosaminoglycan content in knee cartilage: a four-month, randomized, controlled trial in patients at risk of osteoarthritis. Arthritis Rheum. 2005;52:3507–14. - PubMed
-
- Mazzetti I, Magagnoli G, Paoletti S, Uguccioni M, Olivotto E, Vitellozzi R, et al. A role for chemokines in the induction of chondrocyte phenotype modulation. Arthritis Rheum. 2004;50:112–22. - PubMed
-
- Fernandes JC, Martel-Pelletier J, Pelletier JP. The role of cytokines in osteoarthritis pathophysiology. Biorheology. 2002;39:237–46. - PubMed
-
- Lotz M. Cytokines in cartilage injury and repair. Clin Orthop Relat Res. 2001;(391 Suppl):S108–15. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
