

Paired-end mapping reveals extensive structural variation in the human genome
- PMID: 17901297
- PMCID: PMC2674581
- DOI: 10.1126/science.1149504
Paired-end mapping reveals extensive structural variation in the human genome
Abstract
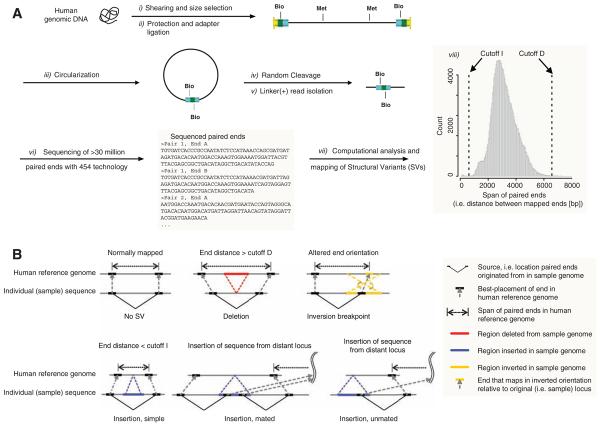
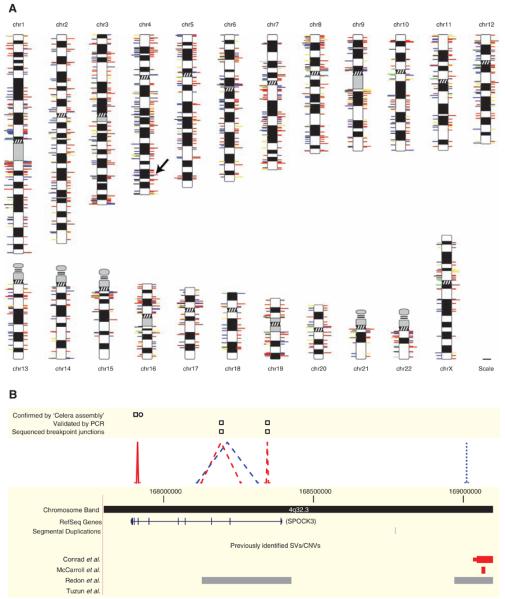
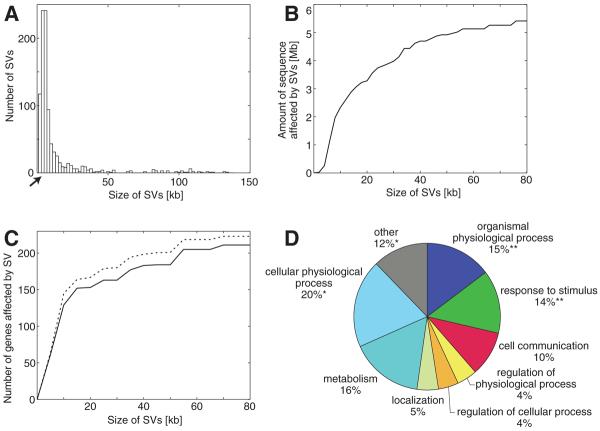
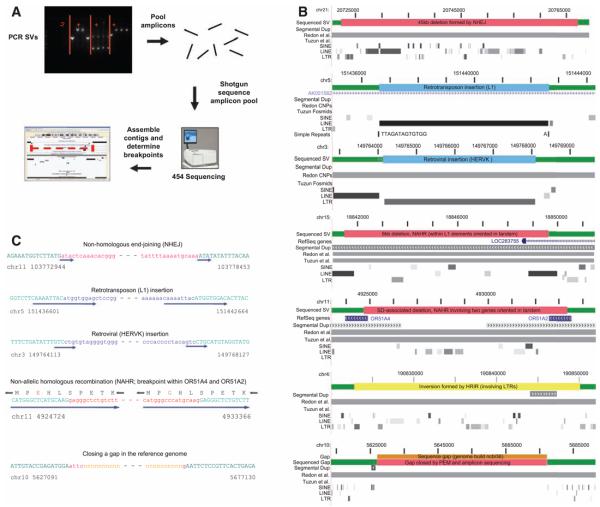
Structural variation of the genome involves kilobase- to megabase-sized deletions, duplications, insertions, inversions, and complex combinations of rearrangements. We introduce high-throughput and massive paired-end mapping (PEM), a large-scale genome-sequencing method to identify structural variants (SVs) approximately 3 kilobases (kb) or larger that combines the rescue and capture of paired ends of 3-kb fragments, massive 454 sequencing, and a computational approach to map DNA reads onto a reference genome. PEM was used to map SVs in an African and in a putatively European individual and identified shared and divergent SVs relative to the reference genome. Overall, we fine-mapped more than 1000 SVs and documented that the number of SVs among humans is much larger than initially hypothesized; many of the SVs potentially affect gene function. The breakpoint junction sequences of more than 200 SVs were determined with a novel pooling strategy and computational analysis. Our analysis provided insights into the mechanisms of SV formation in humans.
Figures

Similar articles
-
The fine-scale architecture of structural variants in 17 mouse genomes.Genome Biol. 2012;13(3):R18. doi: 10.1186/gb-2012-13-3-r18. Genome Biol. 2012. PMID: 22439878 Free PMC article.
-
Structural variation in the chicken genome identified by paired-end next-generation DNA sequencing of reduced representation libraries.BMC Genomics. 2011 Feb 3;12:94. doi: 10.1186/1471-2164-12-94. BMC Genomics. 2011. PMID: 21291514 Free PMC article.
-
An integrative probabilistic model for identification of structural variation in sequencing data.Genome Biol. 2012;13(3):R22. doi: 10.1186/gb-2012-13-3-r22. Genome Biol. 2012. PMID: 22452995 Free PMC article.
-
Disruption of regulatory domains and novel transcripts as disease-causing mechanisms.Bioessays. 2023 Oct;45(10):e2300010. doi: 10.1002/bies.202300010. Epub 2023 Jun 29. Bioessays. 2023. PMID: 37381881 Review.
-
Challenges in studying genomic structural variant formation mechanisms: the short-read dilemma and beyond.Bioessays. 2011 Nov;33(11):840-50. doi: 10.1002/bies.201100075. Epub 2011 Sep 30. Bioessays. 2011. PMID: 21959584 Review.
Cited by
-
De novo and inherited CNVs in MZ twin pairs selected for discordance and concordance on Attention Problems.Eur J Hum Genet. 2012 Oct;20(10):1037-43. doi: 10.1038/ejhg.2012.49. Epub 2012 Apr 11. Eur J Hum Genet. 2012. PMID: 22490988 Free PMC article.
-
Inheritance model introduces differential bias in CNV calls between parents and offspring.Genet Epidemiol. 2012 Jul;36(5):488-98. doi: 10.1002/gepi.21643. Epub 2012 May 24. Genet Epidemiol. 2012. PMID: 22628073 Free PMC article.
-
Characterizing polymorphic inversions in human genomes by single-cell sequencing.Genome Res. 2016 Nov;26(11):1575-1587. doi: 10.1101/gr.201160.115. Epub 2016 Jul 29. Genome Res. 2016. PMID: 27472961 Free PMC article.
-
Systems genetics in "-omics" era: current and future development.Theory Biosci. 2013 Mar;132(1):1-16. doi: 10.1007/s12064-012-0168-x. Epub 2012 Nov 9. Theory Biosci. 2013. PMID: 23138757 Review.
-
Long non-coding RNAs in innate and adaptive immunity.Virus Res. 2016 Jan 2;212:146-60. doi: 10.1016/j.virusres.2015.07.003. Epub 2015 Jul 9. Virus Res. 2016. PMID: 26166759 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
