

Cytochrome c oxidase deficiency in neurons decreases both oxidative stress and amyloid formation in a mouse model of Alzheimer's disease
- PMID: 17715058
- PMCID: PMC1955773
- DOI: 10.1073/pnas.0705738104
Cytochrome c oxidase deficiency in neurons decreases both oxidative stress and amyloid formation in a mouse model of Alzheimer's disease
Abstract
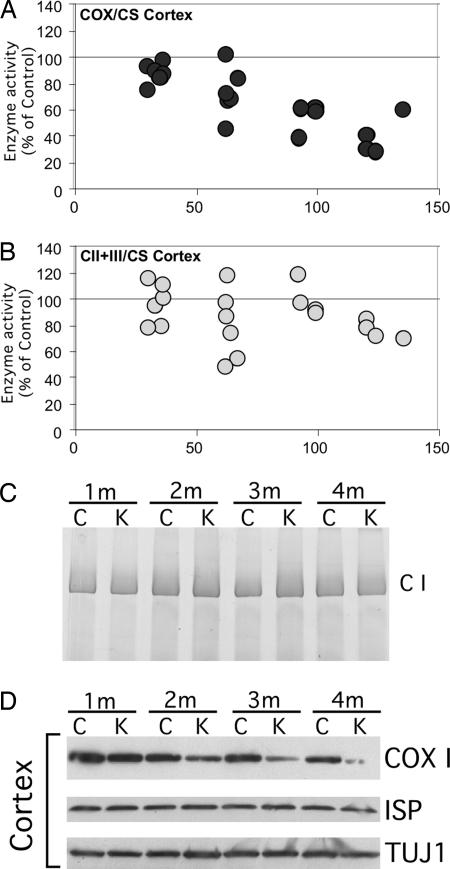
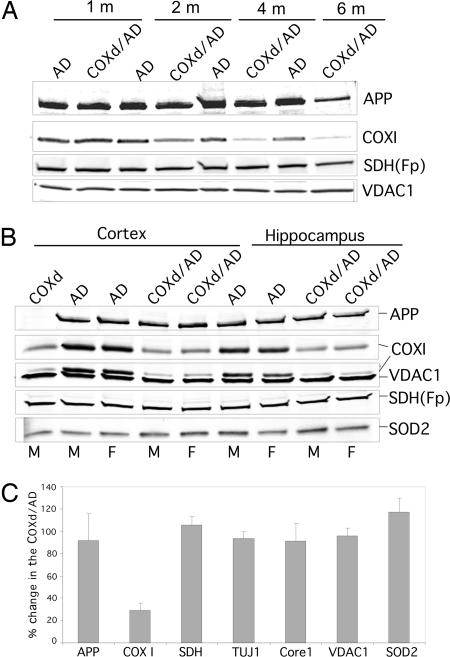
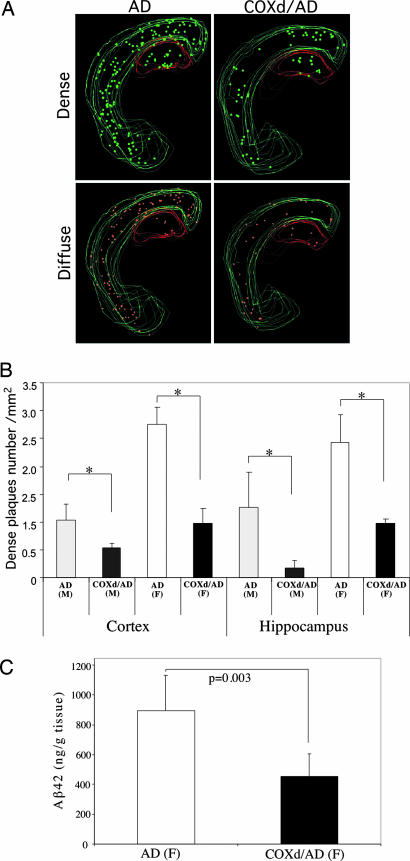
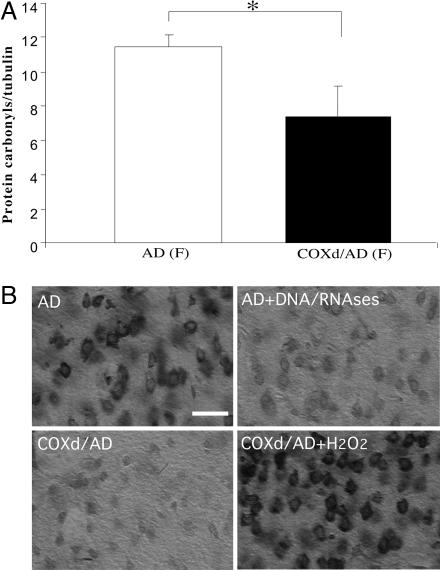
Defects in the mitochondrial cytochrome c oxidase (COX) have been associated with Alzheimer's Disease, in which the age-dependent accumulation of beta-amyloid plays an important role in synaptic dysfunction and neurodegeneration. To test the possibility that age-dependent decline in the mitochondrial respiratory function, especially COX activity, may participate in the formation and accumulation of beta-amyloid, we generated mice expressing mutant amyloid precursor protein and mutant presenilin 1 in a neuron-specific COX-deficient background. A neuron-specific COX-deficient mouse was generated by the Cre-loxP system, in which the COX10 gene was deleted by a CamKIIalpha promoter-driven Cre-recombinase. COX10 is a farnesyltransferase involved in the biosynthesis of heme a, required for COX assembly and function. These KO mice showed an age-dependent COX deficiency in the cerebral cortex and hippocampus. Surprisingly, COX10 KO mice exhibited significantly fewer amyloid plaques in their brains compared with the COX-competent transgenic mice. This reduction in amyloid plaques in the KO mouse was accompanied by a reduction in Abeta42 level, beta-secretase activity, and oxidative damage. Likewise, production of reactive oxygen species from cells with partial COX activity was not elevated. Collectively, our results suggest that, contrary to previous models, a defect in neuronal COX does not increase oxidative damage nor predispose for the formation of amyloidgenic amyloid precursor protein fragments.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Mice lacking COX10 in skeletal muscle recapitulate the phenotype of progressive mitochondrial myopathies associated with cytochrome c oxidase deficiency.Hum Mol Genet. 2005 Sep 15;14(18):2737-48. doi: 10.1093/hmg/ddi307. Epub 2005 Aug 15. Hum Mol Genet. 2005. PMID: 16103131 Free PMC article.
-
Mitochondria and vascular lesions as a central target for the development of Alzheimer's disease and Alzheimer disease-like pathology in transgenic mice.Neurol Res. 2003 Sep;25(6):665-74. doi: 10.1179/016164103101201977. Neurol Res. 2003. PMID: 14503022
-
Regional brain metabolism with cytochrome c oxidase histochemistry in a PS1/A246E mouse model of autosomal dominant Alzheimer's disease: correlations with behavior and oxidative stress.Neurochem Int. 2009 Dec;55(8):806-14. doi: 10.1016/j.neuint.2009.08.005. Epub 2009 Aug 12. Neurochem Int. 2009. PMID: 19682525
-
Dysfunction of mitochondria and oxidative stress in the pathogenesis of Alzheimer's disease: on defects in the cytochrome c oxidase complex and aldehyde detoxification.J Alzheimers Dis. 2006 Jul;9(2):155-66. doi: 10.3233/jad-2006-9208. J Alzheimers Dis. 2006. PMID: 16873963 Review.
-
The role of cytochrome c oxidase deficiency in ROS and amyloid plaque formation.J Bioenerg Biomembr. 2009 Oct;41(5):453-6. doi: 10.1007/s10863-009-9245-3. J Bioenerg Biomembr. 2009. PMID: 19795195 Review.
Cited by
-
Lipodystrophy in methylmalonic acidemia associated with elevated FGF21 and abnormal methylmalonylation.JCI Insight. 2024 Feb 22;9(4):e174097. doi: 10.1172/jci.insight.174097. JCI Insight. 2024. PMID: 38271099 Free PMC article.
-
Amyloid-β peptide binds to cytochrome C oxidase subunit 1.PLoS One. 2012;7(8):e42344. doi: 10.1371/journal.pone.0042344. Epub 2012 Aug 21. PLoS One. 2012. PMID: 22927926 Free PMC article.
-
Ablation of Cytochrome c in Adult Forebrain Neurons Impairs Oxidative Phosphorylation Without Detectable Apoptosis.Mol Neurobiol. 2019 May;56(5):3722-3735. doi: 10.1007/s12035-018-1335-y. Epub 2018 Sep 6. Mol Neurobiol. 2019. PMID: 30191381 Free PMC article.
-
Mitochondrial function and Aβ in Alzheimer's disease postmortem brain.Neurobiol Dis. 2022 Sep;171:105781. doi: 10.1016/j.nbd.2022.105781. Epub 2022 Jun 3. Neurobiol Dis. 2022. PMID: 35667615 Free PMC article.
-
Impaired mitochondrial energy production and ABC transporter function-A crucial interconnection in dementing proteopathies of the brain.Mech Ageing Dev. 2013 Oct;134(10):506-15. doi: 10.1016/j.mad.2013.08.007. Epub 2013 Sep 3. Mech Ageing Dev. 2013. PMID: 24012632 Free PMC article. Review.
References
-
- Hardy J. Neuron. 2006;52:3–13. - PubMed
-
- Chong YH, Shin YJ, Lee EO, Kayed R, Glabe CG, Tenner AJ. J Biol Chem. 2006;281:20315–20325. - PubMed
-
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Nat Neurosci. 2005;8:79–84. - PubMed
-
- Gouras GK, Almeida CG, Takahashi RH. Neurobiol Aging. 2005;26:1235–1244. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
