

Mutations in UPF3B, a member of the nonsense-mediated mRNA decay complex, cause syndromic and nonsyndromic mental retardation
- PMID: 17704778
- PMCID: PMC2872770
- DOI: 10.1038/ng2100
Mutations in UPF3B, a member of the nonsense-mediated mRNA decay complex, cause syndromic and nonsyndromic mental retardation
Abstract
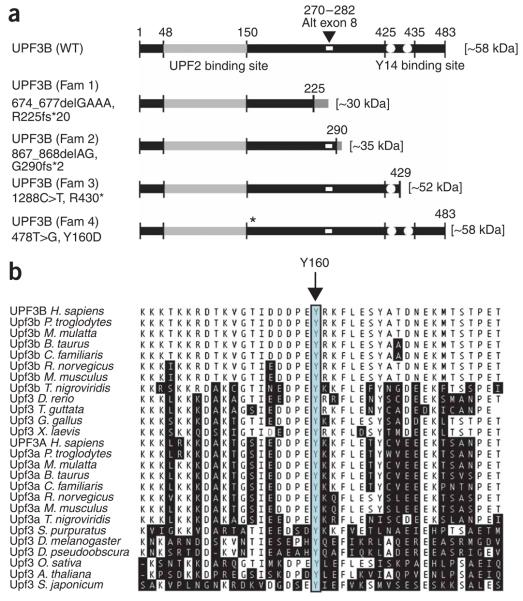
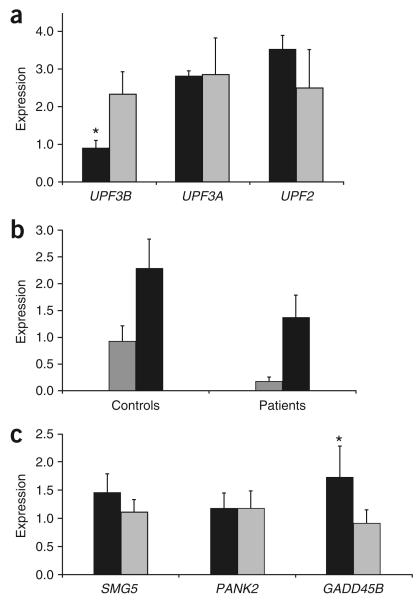
Nonsense-mediated mRNA decay (NMD) is of universal biological significance. It has emerged as an important global RNA, DNA and translation regulatory pathway. By systematically sequencing 737 genes (annotated in the Vertebrate Genome Annotation database) on the human X chromosome in 250 families with X-linked mental retardation, we identified mutations in the UPF3 regulator of nonsense transcripts homolog B (yeast) (UPF3B) leading to protein truncations in three families: two with the Lujan-Fryns phenotype and one with the FG phenotype. We also identified a missense mutation in another family with nonsyndromic mental retardation. Three mutations lead to the introduction of a premature termination codon and subsequent NMD of mutant UPF3B mRNA. Protein blot analysis using lymphoblastoid cell lines from affected individuals showed an absence of the UPF3B protein in two families. The UPF3B protein is an important component of the NMD surveillance machinery. Our results directly implicate abnormalities of NMD in human disease and suggest at least partial redundancy of NMD pathways.
Figures




Similar articles
-
Mutations of the UPF3B gene, which encodes a protein widely expressed in neurons, are associated with nonspecific mental retardation with or without autism.Mol Psychiatry. 2010 Jul;15(7):767-76. doi: 10.1038/mp.2009.14. Epub 2009 Feb 24. Mol Psychiatry. 2010. PMID: 19238151
-
Exome sequencing identifies UPF3B as the causative gene for a Chinese non-syndrome mental retardation pedigree.Clin Genet. 2013 Jun;83(6):560-4. doi: 10.1111/cge.12014. Epub 2012 Sep 28. Clin Genet. 2013. PMID: 22957832
-
A synonymous UPF3B variant causing a speech disorder implicates NMD as a regulator of neurodevelopmental disorder gene networks.Hum Mol Genet. 2020 Aug 29;29(15):2568-2578. doi: 10.1093/hmg/ddaa151. Hum Mol Genet. 2020. PMID: 32667670 Free PMC article.
-
Mechanism, factors, and physiological role of nonsense-mediated mRNA decay.Cell Mol Life Sci. 2015 Dec;72(23):4523-44. doi: 10.1007/s00018-015-2017-9. Epub 2015 Aug 18. Cell Mol Life Sci. 2015. PMID: 26283621 Free PMC article. Review.
-
MicroRNA-mediated regulation of nonsense-mediated mRNA decay factors: Insights into microRNA prediction tools and profiling techniques.Biochim Biophys Acta Gene Regul Mech. 2024 Jun;1867(2):195022. doi: 10.1016/j.bbagrm.2024.195022. Epub 2024 Mar 2. Biochim Biophys Acta Gene Regul Mech. 2024. PMID: 38437914 Review.
Cited by
-
Nonsense-mediated mRNA decay factor Upf2 exists in both the nucleoplasm and the cytoplasm.Mol Med Rep. 2016 Jul;14(1):655-60. doi: 10.3892/mmr.2016.5331. Epub 2016 May 24. Mol Med Rep. 2016. PMID: 27221324 Free PMC article.
-
Contribution of rare and common variants to intellectual disability in a sub-isolate of Northern Finland.Nat Commun. 2019 Jan 24;10(1):410. doi: 10.1038/s41467-018-08262-y. Nat Commun. 2019. PMID: 30679432 Free PMC article.
-
Generation of a Magoh conditional allele in mice.Genesis. 2014 Aug;52(8):752-8. doi: 10.1002/dvg.22788. Epub 2014 May 9. Genesis. 2014. PMID: 24771530 Free PMC article.
-
UPF2, a nonsense-mediated mRNA decay factor, is required for prepubertal Sertoli cell development and male fertility by ensuring fidelity of the transcriptome.Development. 2015 Jan 15;142(2):352-62. doi: 10.1242/dev.115642. Epub 2014 Dec 11. Development. 2015. PMID: 25503407 Free PMC article.
-
Nonsense in the testis: multiple roles for nonsense-mediated decay revealed in male reproduction.Biol Reprod. 2017 May 1;96(5):939-947. doi: 10.1093/biolre/iox033. Biol Reprod. 2017. PMID: 28444146 Free PMC article. Review.
References
-
- Maquat LE. Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat. Rev. Mol. Cell Biol. 2004;5:89–99. - PubMed
-
- Conti E, Izaurralde E. Nonsense-mediated mRNA decay: molecular insights and mechanistic variations across species. Curr. Opin. Cell Biol. 2005;17:316–325. - PubMed
-
- Medghalchi SM, et al. Rent1, a trans-effector of nonsense-mediated mRNA decay, is essential for mammalian embryonic viability. Hum. Mol. Genet. 2001;10:99–105. - PubMed
-
- Weischenfeldt J, Lykke-Andersen J, Porse B. Messenger RNA surveillance: neutralising natural nonsense. Curr. Biol. 2005;15:R559–R562. - PubMed
-
- Lujan JE, Carlin ME, Lubs HA. A form of X-linked mental retardation with Marfanoid habitus. Am. J. Med. Genet. 1984;17:311–322. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
