

Opposite regulation of oligodendrocyte apoptosis by JNK3 and Pin1 after spinal cord injury
- PMID: 17670986
- PMCID: PMC3401937
- DOI: 10.1523/JNEUROSCI.2478-07.2007
Opposite regulation of oligodendrocyte apoptosis by JNK3 and Pin1 after spinal cord injury
Abstract
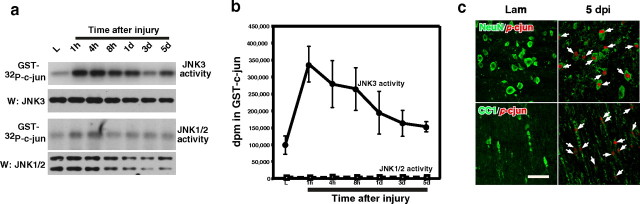
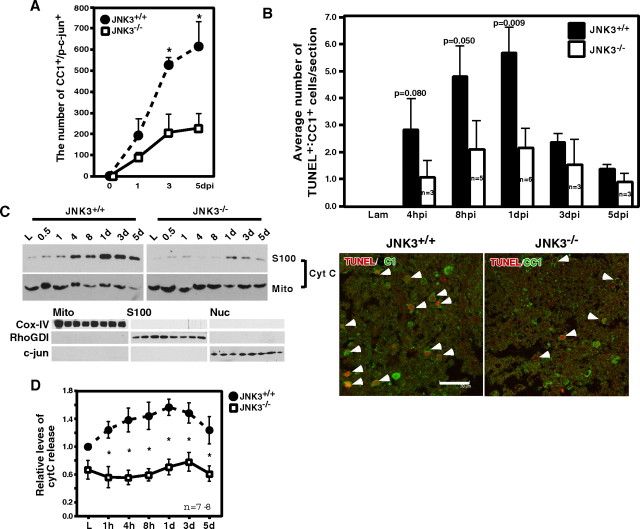
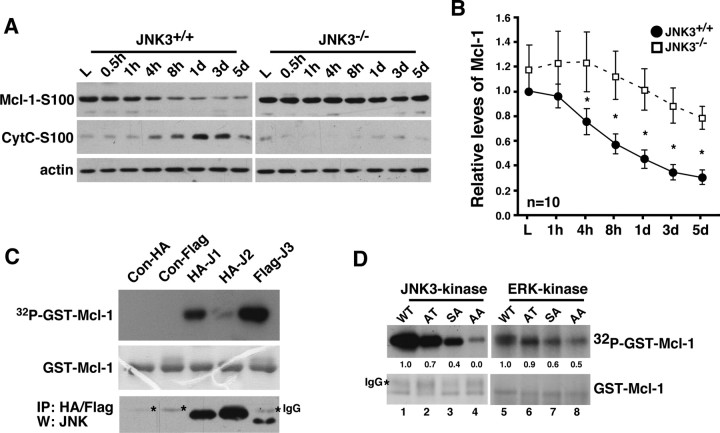
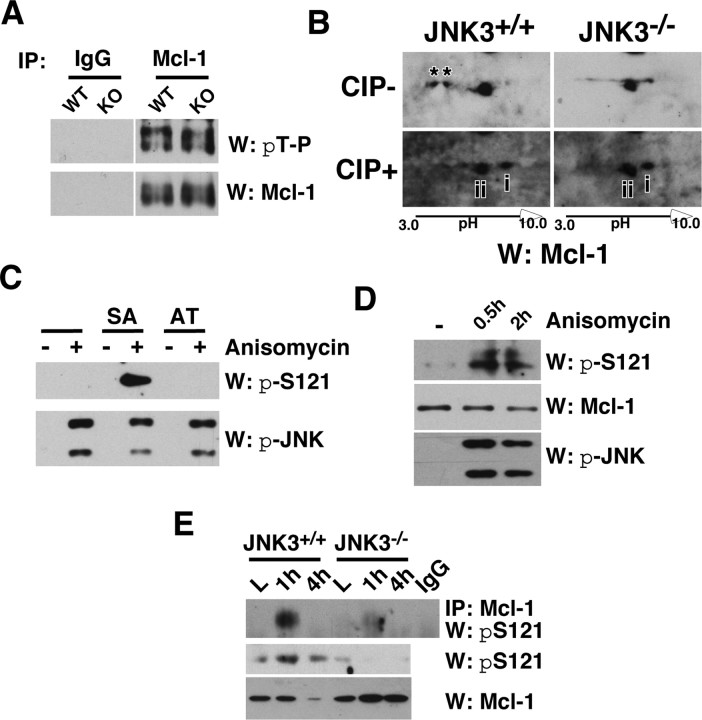
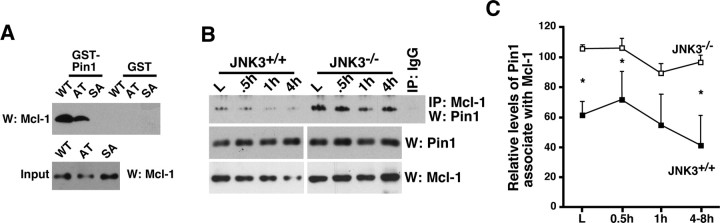
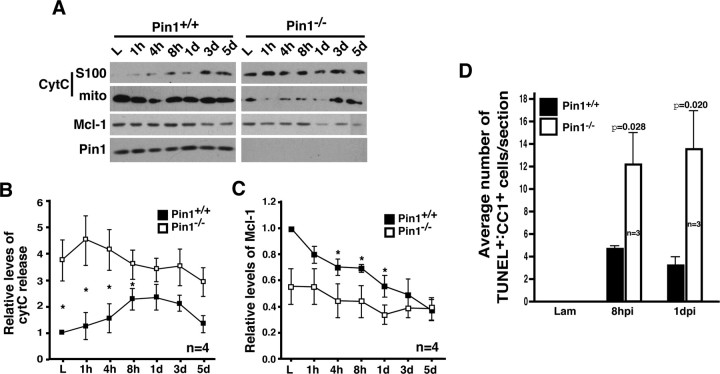
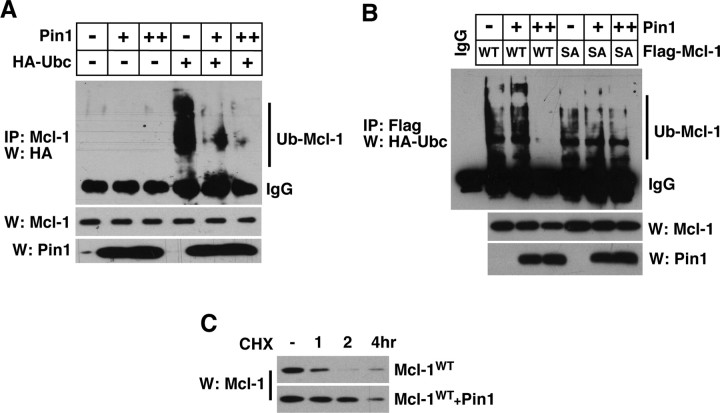
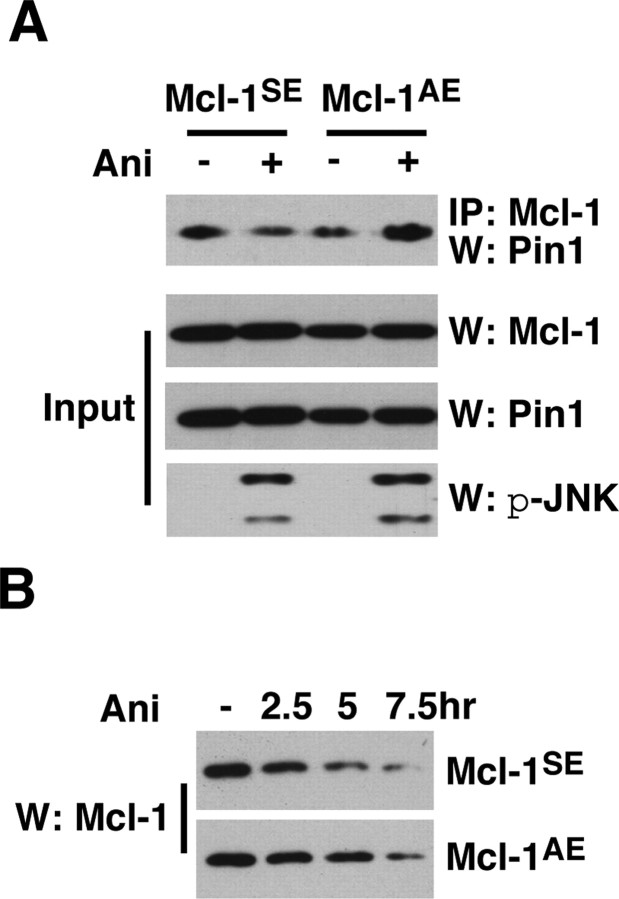
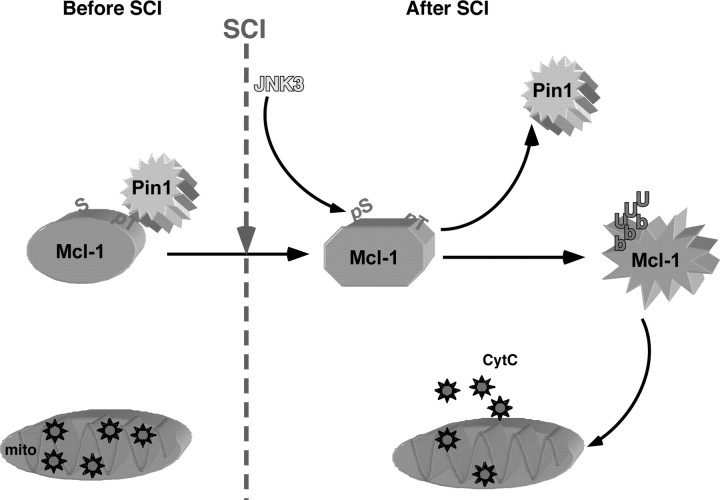
Although oligodendrocytes undergo apoptosis after spinal cord injury, molecular mechanisms responsible for their death have been unknown. We report that oligodendrocyte apoptosis is regulated oppositely by c-Jun N-terminal kinase 3 (JNK3) and protein interacting with the mitotic kinase, never in mitosis A I (Pin1), the actions of which converge on myeloid cell leukemia sequence-1 (Mcl-1). Activated after injury, JNK3 induces cytochrome c release by facilitating the degradation of Mcl-1, the stability of which is maintained in part by Pin1. Pin1 binds Mcl-1 at its constitutively phosphorylated site, Thr163Pro, and stabilizes it by inhibiting ubiquitination. After injury JNK3 phosphorylates Mcl-1 at Ser121Pro, facilitating the dissociation of Pin1 from Mcl-1. JNK3 thus induces Mcl-1 degradation by counteracting the protective binding of Pin1. These results are confirmed by the opposing phenotypes observed between JNK3-/- and Pin1-/- mice: oligodendrocyte apoptosis and cytochrome c release are reduced in JNK3-/- but elevated in Pin1-/- mice. This report thus unveils a mechanism by which cytochrome c release is under the opposite control of JNK3 and Pin1, regulators for which the activities are intricately coupled.
Figures
Similar articles
-
17β-Estradiol inhibits apoptotic cell death of oligodendrocytes by inhibiting RhoA-JNK3 activation after spinal cord injury.Endocrinology. 2012 Aug;153(8):3815-27. doi: 10.1210/en.2012-1068. Epub 2012 Jun 14. Endocrinology. 2012. PMID: 22700771
-
Deletion of the c-Jun N-terminal kinase 3 gene protects neonatal mice against cerebral hypoxic-ischaemic injury.J Cereb Blood Flow Metab. 2007 May;27(5):1022-32. doi: 10.1038/sj.jcbfm.9600413. Epub 2006 Oct 25. J Cereb Blood Flow Metab. 2007. PMID: 17063149
-
c-Jun N-terminal kinase induces axonal degeneration and limits motor recovery after spinal cord injury in mice.Neurosci Res. 2011 Nov;71(3):266-77. doi: 10.1016/j.neures.2011.07.1830. Epub 2011 Jul 28. Neurosci Res. 2011. PMID: 21824499
-
The peptidyl-prolyl isomerase Pin1.Prog Cell Cycle Res. 2003;5:477-87. Prog Cell Cycle Res. 2003. PMID: 14593743 Review.
-
[Recent advances in the study of pin1 and its inhibitors].Yao Xue Xue Bao. 2008 Jan;43(1):9-17. Yao Xue Xue Bao. 2008. PMID: 18357725 Review. Chinese.
Cited by
-
Analgesic effect of acupuncture is mediated via inhibition of JNK activation in astrocytes after spinal cord injury.PLoS One. 2013 Sep 9;8(9):e73948. doi: 10.1371/journal.pone.0073948. eCollection 2013. PLoS One. 2013. PMID: 24040124 Free PMC article.
-
Advances of the MAPK pathway in the treatment of spinal cord injury.CNS Neurosci Ther. 2024 Jun;30(6):e14807. doi: 10.1111/cns.14807. CNS Neurosci Ther. 2024. PMID: 38887853 Free PMC article. Review.
-
The peptidyl-prolyl isomerase Pin1 up-regulation and proapoptotic function in dopaminergic neurons: relevance to the pathogenesis of Parkinson disease.J Biol Chem. 2013 Jul 26;288(30):21955-71. doi: 10.1074/jbc.M112.444224. Epub 2013 Jun 10. J Biol Chem. 2013. PMID: 23754278 Free PMC article.
-
Effects of hyperbaric oxygen on glucose-regulated protein 78 and c-Jun N-terminal kinase expression after spinal cord injury in rats.Int J Clin Exp Med. 2015 Mar 15;8(3):3309-17. eCollection 2015. Int J Clin Exp Med. 2015. PMID: 26064220 Free PMC article.
-
Amino Acid Composition of a Chum Salmon (Oncorhynchus keta) Skin Gelatin Hydrolysate and Its Antiapoptotic Effects on Etoposide-Induced Osteoblasts.Foods. 2023 Jun 20;12(12):2419. doi: 10.3390/foods12122419. Foods. 2023. PMID: 37372630 Free PMC article.
References
-
- Becker EB, Bonni A. Pin1 mediates neural-specific activation of the mitochondrial apoptotic machinery. Neuron. 2006;49:655–662. - PubMed
-
- Behrens A, Sibilia M, Wagner EF. Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat Genet. 1999;21:326–329. - PubMed
-
- Benn SC, Woolf CJ. Adult neuron survival strategies—slamming on the brakes. Nat Rev Neurosci. 2004;5:686–700. - PubMed
-
- Brecht S, Kirchhof R, Chromik A, Willesen M, Nicolaus T, Raivich G, Wessig J, Waetzig V, Goetz M, Claussen M, Pearse D, Kuan CY, Vaudano E, Behrens A, Wagner E, Flavell RA, Davis RJ, Herdegen T. Specific pathophysiological functions of JNK isoforms in the brain. Eur J Neurosci. 2005;21:363–377. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous