

Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation
- PMID: 17664422
- PMCID: PMC1941831
- DOI: 10.1073/pnas.0701953104
Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation
Abstract
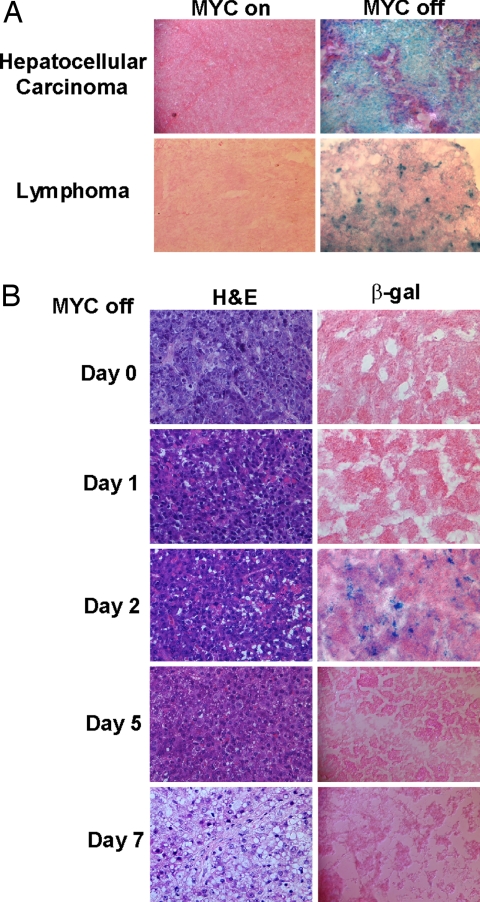
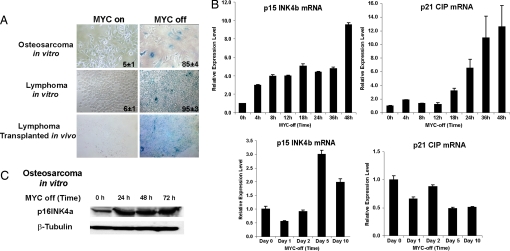
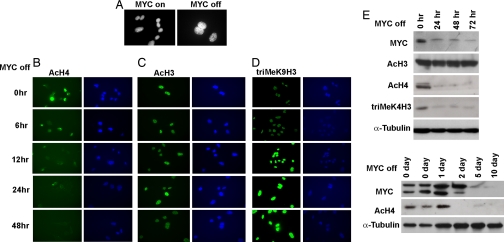
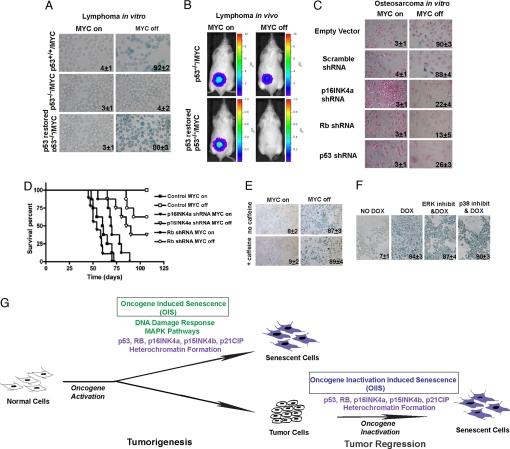
Oncogene-induced senescence is an important mechanism by which normal cells are restrained from malignant transformation. Here we report that the suppression of the c-Myc (MYC) oncogene induces cellular senescence in diverse tumor types including lymphoma, osteosarcoma, and hepatocellular carcinoma. MYC inactivation was associated with prototypical markers of senescence, including acidic beta-gal staining, induction of p16INK4a, and p15INK4b expression. Moreover, MYC inactivation induced global changes in chromatin structure associated with the marked reduction of histone H4 acetylation and increased histone H3 K9 methylation. Osteosarcomas engineered to be deficient in p16INK4a or Rb exhibited impaired senescence and failed to exhibit sustained tumor regression upon MYC inactivation. Similarly, only after lymphomas were repaired for p53 expression did MYC inactivation induce robust senescence and sustained tumor regression. The pharmacologic inhibition of signaling pathways implicated in oncogene-induced senescence including ATM/ATR and MAPK did not prevent senescence associated with MYC inactivation. Our results suggest that cellular senescence programs remain latently functional, even in established tumors, and can become reactivated, serving as a critical mechanism of oncogene addiction associated with MYC inactivation.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
p19ARF is a critical mediator of both cellular senescence and an innate immune response associated with MYC inactivation in mouse model of acute leukemia.Oncotarget. 2015 Feb 28;6(6):3563-77. doi: 10.18632/oncotarget.2969. Oncotarget. 2015. PMID: 25784651 Free PMC article.
-
Sustained regression of tumors upon MYC inactivation requires p53 or thrombospondin-1 to reverse the angiogenic switch.Proc Natl Acad Sci U S A. 2006 Oct 31;103(44):16266-71. doi: 10.1073/pnas.0608017103. Epub 2006 Oct 20. Proc Natl Acad Sci U S A. 2006. PMID: 17056717 Free PMC article.
-
Defects in TGF-beta signaling overcome senescence of mouse keratinocytes expressing v-Ha-ras.Oncogene. 2000 Mar 23;19(13):1698-709. doi: 10.1038/sj.onc.1203471. Oncogene. 2000. PMID: 10763827
-
Tipping the balance: Cdk2 enables Myc to suppress senescence.Cancer Res. 2010 Sep 1;70(17):6687-91. doi: 10.1158/0008-5472.CAN-10-1383. Epub 2010 Aug 16. Cancer Res. 2010. PMID: 20713526 Review.
-
The Myc/macrophage tango: oncogene-induced senescence, Myc style.Semin Cancer Biol. 2011 Dec;21(6):377-84. doi: 10.1016/j.semcancer.2011.10.002. Epub 2011 Oct 13. Semin Cancer Biol. 2011. PMID: 22019769 Review.
Cited by
-
Therapeutic Targeting of MYC in Head and Neck Squamous Cell Carcinoma.Oncoimmunology. 2022 Sep 30;11(1):2130583. doi: 10.1080/2162402X.2022.2130583. eCollection 2022. Oncoimmunology. 2022. PMID: 36211811 Free PMC article.
-
Cellular senescence in cancer: from mechanisms to detection.Mol Oncol. 2021 Oct;15(10):2634-2671. doi: 10.1002/1878-0261.12807. Epub 2020 Oct 22. Mol Oncol. 2021. PMID: 32981205 Free PMC article. Review.
-
Pathways of oncogene-induced senescence in human melanocytic cells.Cell Cycle. 2010 Jul 15;9(14):2782-8. doi: 10.4161/cc.9.14.12551. Epub 2010 Jul 3. Cell Cycle. 2010. PMID: 20676024 Free PMC article. Review. No abstract available.
-
Nuclear Nestin deficiency drives tumor senescence via lamin A/C-dependent nuclear deformation.Nat Commun. 2018 Sep 6;9(1):3613. doi: 10.1038/s41467-018-05808-y. Nat Commun. 2018. PMID: 30190500 Free PMC article.
-
Glioma malignancy is linked to interdependent and inverse AMOG and L1 adhesion molecule expression.BMC Cancer. 2019 Sep 12;19(1):911. doi: 10.1186/s12885-019-6091-5. BMC Cancer. 2019. PMID: 31510944 Free PMC article.
References
-
- Dang CV, O'Donnell KA, Zeller KI, Nguyen T, Osthus RC, Li F. Semin Cancer Biol. 2006;16:253–264. - PubMed
-
- Oster SK, Ho CS, Soucie EL, Penn LZ. Adv Cancer Res. 2002;84:81–154. - PubMed
-
- Secombe J, Pierce SB, Eisenman RN. Cell. 2004;117:153–156. - PubMed
-
- Adhikary S, Eilers M. Nat Rev Mol Cell Biol. 2005;6:635–645. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
