

doi: 10.1083/jcb.200608033.
An ACAP1-containing clathrin coat complex for endocytic recycling
Affiliations
- PMID: 17664335
- PMCID: PMC2064835
- DOI: 10.1083/jcb.200608033
Item in Clipboard
An ACAP1-containing clathrin coat complex for endocytic recycling
J Cell Biol.
.
Abstract
Whether coat proteins play a widespread role in endocytic recycling remains unclear. We find that ACAP1, a GTPase-activating protein (GAP) for ADP-ribosylation factor (ARF) 6, is part of a novel clathrin coat complex that is regulated by ARF6 for endocytic recycling in two key physiological settings, stimulation-dependent recycling of integrin that is critical for cell migration and insulin-stimulated recycling of glucose transporter type 4 (Glut4), which is required for glucose homeostasis. These findings not only advance a basic understanding of an early mechanistic step in endocytic recycling but also shed key mechanistic insights into major physiological events for which this transport plays a critical role.
Figures
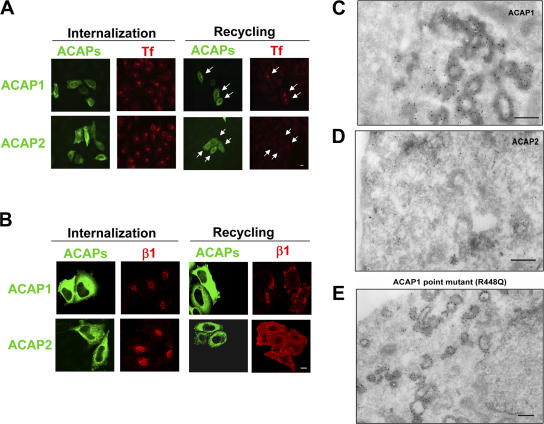
Overexpression of ACAP1 inhibits endocytic recycling and induces membrane coating. (A) Overexpression of ACAP1 but not ACAP2 inhibits TfR recycling from the pericentriolar recycling endosome but not its internalization to this compartment. TRVb-1 cells were transiently transfected with either Flag-ACAP1 or Flag-ACAP2 and assessed for the uptake and recycling of Alexa 594–conjugated transferrin by immunofluorescence microscopy. ACAPs are show in green and transferrin in red. Bar, 15 μm. Arrows indicate cells with high expression of transfected construct. (B) Integrin recycling is similarly affected by ACAP1 overexpression. HeLa cells were transiently transfected with Flag-ACAP1 or Flag-ACAP2 and then antibody-bound surface integrin and tracked for its internalization followed by recycling by immunofluorescence microscopy. ACAPs are shown in green and integrin in red. Bar, 10 μm. (C–E) Overexpression of ACAP1 or its catalytic-dead mutant induces coated membrane structures. HeLa cells were transiently transfected with Flag-tagged ACAP1 (C), ACAP2 (D), or catalytic-dead mutant of ACAP1 (E) and fixed for immunogold EM using anti-Flag antibody. Bars, 200 nm.

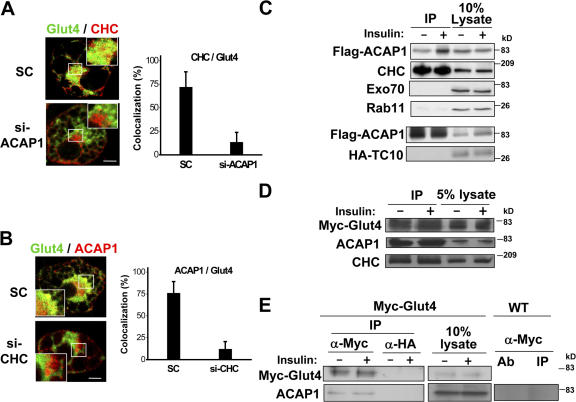
Endosomal clathrin associates with ACAP1 and is required for integrin recycling. (A) Endogenous CHC shows partial colocalization with transfected ACAP1. HeLa cells were transiently transfected with GFP-ACAP1 and examined by immunofluorescence microscopy for ACAP1 (green) and CHC (red). Bar, 10 μm. (B) Clathrin interacts with ACAP1. HeLa cells were transiently transfected with ACAP1-Myc, either permeabilized (to release cytosol) or not, and lysed for immunoprecipitation followed by blotting proteins as indicated. (C) Clathrin can interact directly with ACAP1. ACAP1 as a GST fusion protein was bound to beads and incubated with soluble clathrin triskelia for a pull-down experiment. CHC was detected by immunoblotting for CHC, whereas GST proteins were detected by Coomassie staining. Arrow indicates the position of full-length GST-ACAP1. (D) Knocking down CHC does not prevent the internal accumulation of surface integrin β1. HeLa cells were treated with siRNA against CHC. (top) Cell lysates from different conditions of siRNA treatment (SC denotes scrambled siRNA) were blotted for proteins as indicated. (bottom) Antibody-bound integrin was allowed to accumulate internally and then examined by immunofluorescence microscopy. Integrin is shown in red and clathrin in green. Bar, 10 μm. (E) Knocking down CHC inhibits integrin recycling. A biochemical assay for integrin recycling was performed, with the graph showing the level of recycling integrin remaining internal at the times indicated. The mean from three independent experiments is shown with standard error.

Both ACAP1 and clathrin participate in Glut4 recycling in adipocytes. (A) Localization of different proteins in adipocytes. Differentiated 3T3-L1 cells at the basal condition were examined by confocal microscopy for (top row) ACAP1 (green) and CHC (red), (second row) HA-Glut4-GFP (green) and CHC (red), (third row) HA-Glut4-GFP (green) and ACAP1 (pseudored), (fourth row) sortilin-6xHis (green) and ACAP1 (red), and (bottom row) ACAP1 (green) and syntaxin 6 (red). Bars, 10 μm. Note that staining for ACAP1, CHC, and syntaxin 6 involved endogenous proteins. (B) Knocking down ACAP1 inhibits insulin-stimulated redistribution of Glut4. Differentiated 3T3-L1 cells stably expressing HA-Glut4-GFP were treated with siRNA conditions as indicated. (top) Cell lysates were immunoblotted for proteins as indicated. (bottom) Cells were assayed for Glut4 translocation by measuring the level of surface Glut4 (tracked by HA antibody binding to unpermeabilized cells) when normalized to total Glut4 (indicated by GFP intensity), comparing basal versus stimulated condition. The mean from examination of 10 randomly selected cells is shown with standard error. (C) Knocking down ACAP1 also inhibits glucose uptake. Differentiated 3T3-L1 cells were treated with distinct siRNAs against ACAP1 and assessed by the cellular glucose uptake assay. The mean from three independent experiments is shown with standard error. (D) Knocking down CHC also inhibits insulin-stimulated redistribution of Glut4. The same experiment as described in B was performed, except cells were treated with siRNA against CHC.
Interaction between ACAP1 and CHC. (A) Knocking down ACAP1 reduces the colocalization of CHC with internal Glut4. Differentiated 3T3-L1 cells stably expressing HA-Glut4-GFP were treated with siRNA against ACAP1 and examined at the basal condition by confocal microscopy for Glut4 (green) and CHC (red). Bar, 10 μm. 10 cells were also randomly selected and quantified for the fraction of Glut4 that colocalized with CHC. The mean with standard error is shown. (B) Knocking down CHC also reduces the colocalization of ACAP1 with internal Glut4. The same experiment as described in A was performed, except that cells treated with siRNA against CHC and the degree of colocalization between Glut4 (green) and ACAP1 (red) was assessed. (C) ACAP1 interacts with CHC. Differentiated 3T3-L1 cells transfected with Flag-ACAP1 and treated with conditions of stimulation as noted were lysed followed by immunoprecipitation using an anti-Flag antibody and immunoblotting for proteins as indicated. TC10 was detected by transfecting cells with HA-TC10. (D) Both ACAP1 and CHC are in a complex with Glut4. Differentiated 3T3-L1 cells stably expressing Myc-Glut4 were analyzed in a coprecipitation experiment as described in C. (E) Specificity controls for immunoprecipitating Myc-Glut4. Experiment described in D was expanded to include additional controls as indicated.
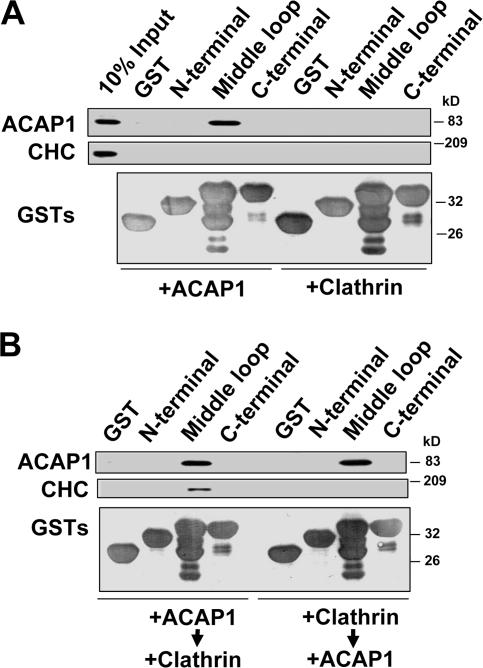
Determining how ACAP1 and CHC interact with Glut4. (A) The middle cytoplasmic loop of Glut4 interacts directly with ACAP1. The different cytoplasmic domains of Glut4 as indicated were bound to beads as GST fusion proteins and incubated with either soluble ACAP1 or clathrin triskelia. Bound soluble components were detected by immunoblotting, whereas GST proteins were detected by Coomassie staining. (B) ACAP1 bridges the binding of clathrin triskelia to Glut4. The same pull-down experiment was performed as in A, except that soluble proteins were incubated sequentially as indicated. Note that unbound soluble proteins after the first incubation were removed before the second incubation.
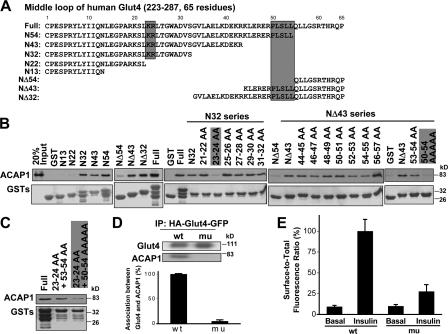
Key residues in Glut4 that mediate its direct binding to ACAP1 define recycling sorting signals. (A) A scheme showing the sequence of the middle domain of Glut4, with truncation constructs as noted. Critical residues that define recycling sorting signals are boxed. (B) Systematic mutagenesis of the middle domain to identify key residues within two distinct regions that mediate binding to ACAP1. Different constructs as noted were generated as GST fusions and incubated with ACAP1 in pull-down experiments. Truncation mutants were initially screened (left), followed by alanine-scanning mutagenesis of two regions identified to bind ACAP1 (middle and right). ACAP1 was detected by immunoblotting, whereas GST fusions were detected by Coomassie staining. (C) Mutation of key residues within two distinct regions of the entire middle domain reduces its binding to ACAP1. Experiments similar to that described in B were performed using different GST fusions as noted. (D) Mutation of key residues in Glut4 prevents its interaction with ACAP1 in vivo. Wild-type or a mutant form of HA-Glut4-GFP (generated by replacing positions 23, 24, and 50–54 in the middle cytoplasmic domain, as defined in A, to alanines) was transfected intodifferentiated 3T3-L1 cells. Cell lysates were immunoprecipitated for the chimeric Glut4 followed by immunoblotting for ACAP1. The mean from three experiments with standard error is also shown. (E) Key residues in Glut4 that mediate its binding to ACAP1 represent recycling sorting signals. Wild-type or mutant form of HA-Glut4-GFP (as described in D) were transfected into differentiated 3T3-L1 cells and assayed for their translocation by measuring their surface level (tracked by HA antibody binding to unpermeabilized cells) when normalized to their total (indicated by GFP intensity), comparing basal versus stimulated condition. The mean from examination of 10 randomly selected cells is shown with standard error.
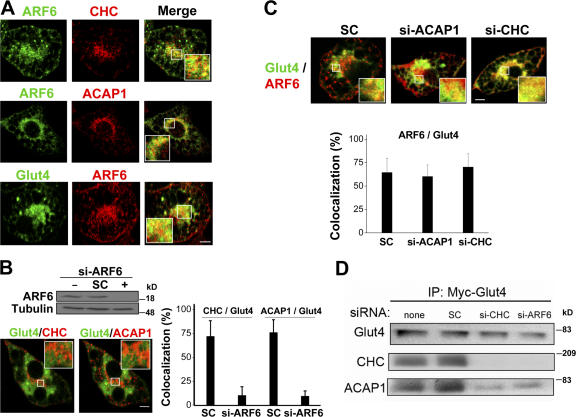
Regulation of the novel clathrin coat complex by ARF6. (A) Endogenous ARF6 shows considerable colocalization with endogenous ACAP1 and CHC and with internal Glut4 in differentiated 3T3-L1 cells. Differentiated 3T3-L1 cells were examined under the basal condition by confocal microscopy for the following combinations: (top) ARF6 (green) and CHC (red), (middle) ARF6 (green) and ACAP1 (red), and (bottom) ARF6 (red) and Glut4 (green). Bar, 10 μm. (B) Knocking down ARF6 reduces the colocalization of both ACAP1 and CHC with internal Glut4. Differentiated 3T3-L1 cells stably expressing HA-Glut4-GFP were treated with siRNA against ARF6. (top) Cells were blotted for proteins as indicated. (bottom) Cells were also examined at the basal condition by confocal microscopy for Glut4 (green) and CHC (red), or Glut4 (green) and ACAP1 (pseudored). Bar, 10 μm. The degree of colocalization derived from examining 10 randomly selected cells was also quantified. (C) Knocking down either ACAP1 or CHC does not affect the colocalization of ARF6 and internal Glut4. Differentiated 3T3-L1 cells stably expressing HA-Glut4-GFP were treated with siRNA against either ACAP1 or CHC and examined at the basal condition by confocal microscopy for Glut4 (green) and ARF6 (red). Bar, 10 μm. The degree of colocalization derived from examining 10 randomly selected cells was also quantified. (D) Knocking down either CHC or ARF6 disrupts the association of Glut4 with ACAP1 and CHC. Cell lysates derived from differentiated 3T3-L1 cells stably expressing Myc-Glut4 were immunoprecipitated using the anti-Myc antibody followed by immunoblotting for proteins as indicated.
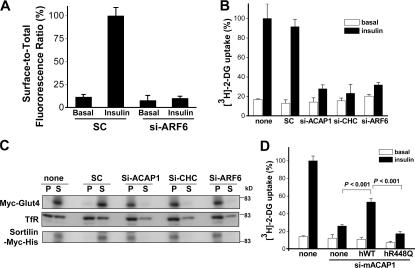
Characterizing the novel coat complex regulated by ARF6 in Glut4 recycling. (A) Knocking down ARF6 inhibits insulin-stimulated redistribution of Glut4. Differentiated 3T3-L1 cells transfected with HA-Glut4-GFP were treated with different siRNA conditions as noted and assayed for Glut4 translocation by measuring the level of surface Glut4 (tracked by HA antibody binding to unpermeabilized cells) when normalized to total Glut4 (indicated by GFP intensity), comparing basal versus stimulated condition. The mean from examination of 10 randomly selected cells is shown with standard error. (B) Silencing ACAP1, CHC, or ARF6 leads to similar levels of reduction in the cellular uptake of glucose. Differentiated 3T3-L1 cells were treated with different siRNA conditions as noted and assayed for the uptake of radioactive 2-deoxy-glucose. The mean from three independent experiments is shown with standard error. (C) Silencing ACAP1, CHC, or ARF6 redistributes Glut4, sortilin, and TfR from vesicular membranes to compartmental membranes. Differentiated 3T3-L1 cells stably expressing both myc-Glut4 and sortilin-myc-his were homogenized and subjected to centrifugation to derive pellet that contains compartmental membrane and supernatant that contains vesicular membrane and cytosol. Bothfractions were immunoblotted for endosomal proteins as indicated to reveal their relative distribution in compartmental and vesicular membranes. (D) GAP activity of ACAP1 plays a role in Glut4 recycling. 3T3-L1 cells were stably transfected with either human wild-type or mutant (R448Q) ACAP1. After differentiation, cells were silenced for endogenous mouse ACAP1 and assayed for the cellular uptake of glucose as described in B.
Similar articles
-
An ACAP1 coat complex acting in endocytic recycling.Methods Cell Biol. 2015;130:81-99. doi: 10.1016/bs.mcb.2015.03.019. Epub 2015 Jun 11. Methods Cell Biol. 2015. PMID: 26360030
-
A novel GTPase-activating protein for ARF6 directly interacts with clathrin and regulates clathrin-dependent endocytosis.Mol Biol Cell. 2005 Apr;16(4):1617-28. doi: 10.1091/mbc.e04-08-0683. Epub 2005 Jan 19. Mol Biol Cell. 2005. PMID: 15659652 Free PMC article.
-
Mechanistic insights into regulated cargo binding by ACAP1 protein.J Biol Chem. 2012 Aug 17;287(34):28675-85. doi: 10.1074/jbc.M112.378810. Epub 2012 May 29. J Biol Chem. 2012. PMID: 22645133 Free PMC article.
-
Arf GAPs: A family of proteins with disparate functions that converge on a common structure, the integrin adhesion complex.Small GTPases. 2019 Jul;10(4):280-288. doi: 10.1080/21541248.2017.1299271. Epub 2017 Mar 31. Small GTPases. 2019. PMID: 28362242 Free PMC article. Review.
-
GRAF1-dependent endocytosis.Biochem Soc Trans. 2009 Oct;37(Pt 5):1061-5. doi: 10.1042/BST0371061. Biochem Soc Trans. 2009. PMID: 19754452 Review.
Cited by
-
Arf GAPs and molecular motors.Small GTPases. 2019 May;10(3):196-209. doi: 10.1080/21541248.2017.1308850. Epub 2017 Apr 21. Small GTPases. 2019. PMID: 28430047 Free PMC article. Review.
-
Akt may associate with insulin-responsive vesicles via interaction with sortilin.FEBS Lett. 2024 Feb;598(4):390-399. doi: 10.1002/1873-3468.14790. Epub 2023 Dec 21. FEBS Lett. 2024. PMID: 38105115
-
Clathrin and AP1B: key roles in basolateral trafficking through trans-endosomal routes.FEBS Lett. 2009 Dec 3;583(23):3784-95. doi: 10.1016/j.febslet.2009.10.050. Epub 2009 Oct 23. FEBS Lett. 2009. PMID: 19854182 Free PMC article. Review.
-
Exclusion of integrins from CNS axons is regulated by Arf6 activation and the AIS.J Neurosci. 2015 May 27;35(21):8359-75. doi: 10.1523/JNEUROSCI.2850-14.2015. J Neurosci. 2015. PMID: 26019348 Free PMC article.
-
RGD-Binding Integrins Revisited: How Recently Discovered Functions and Novel Synthetic Ligands (Re-)Shape an Ever-Evolving Field.Cancers (Basel). 2021 Apr 4;13(7):1711. doi: 10.3390/cancers13071711. Cancers (Basel). 2021. PMID: 33916607 Free PMC article. Review.
References
-
- Balañá, M.E., F. Niedergang, A. Subtil, A. Alcover, P. Chavrier, and A. Daultry-Varsat. 2005. ARF6 GTPase controls bacterial invasion by actin remodelling. J. Cell Sci. 118:2201–2210. - PubMed
-
- Barlowe, C., L. Orci, T. Yeung, M. Hosobuchi, S. Hamamoto, N. Salama, M.F. Rexach, M. Ravazzola, M. Amherdt, and R. Schekman. 1994. COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell. 77:895–907. - PubMed
-
- Bogan, J.S., N. Hendon, A.E. McKee, T.S. Tsao, and H.F. Lodish. 2003. Functional cloning of TUG as a regulator of GLUT4 glucose transporter trafficking. Nature. 425:727–733. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
