

Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility
- PMID: 17639084
- PMCID: PMC1920173
- DOI: 10.1101/gad.1563807
Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility
Abstract
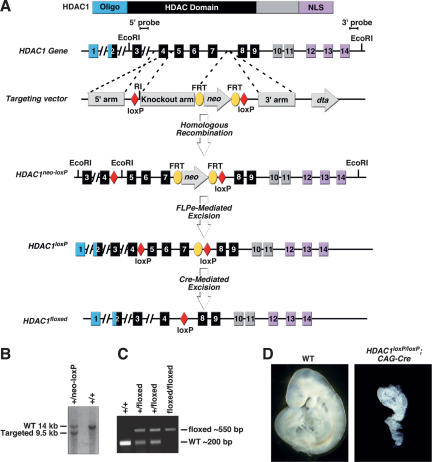
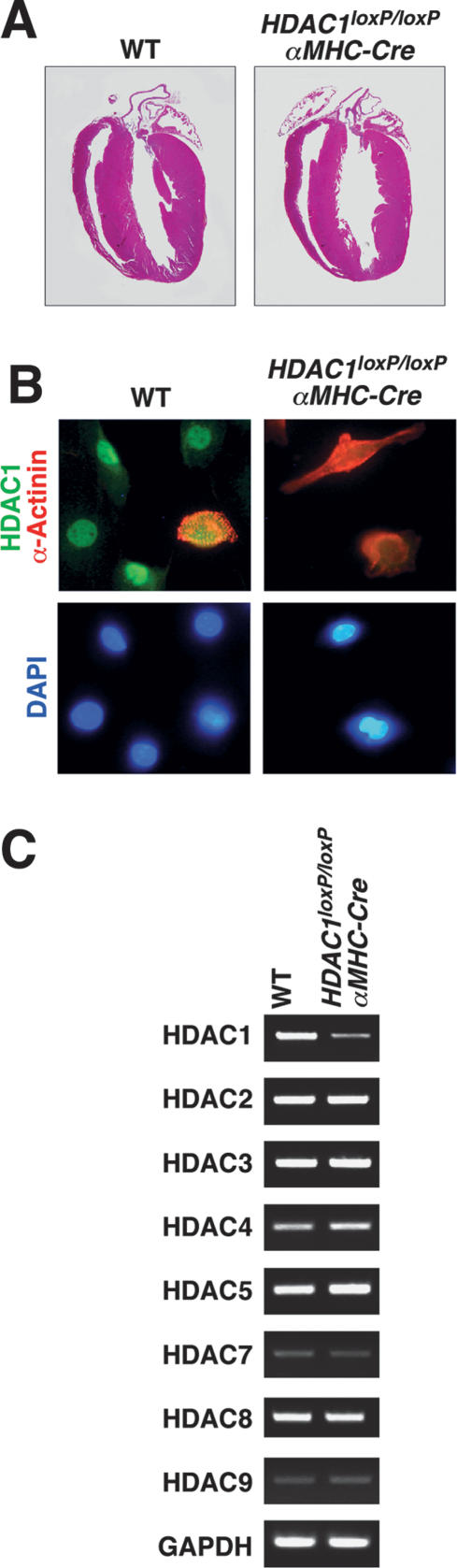
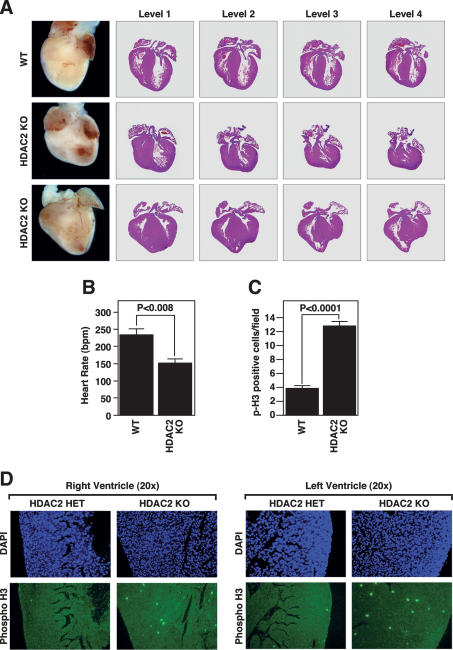
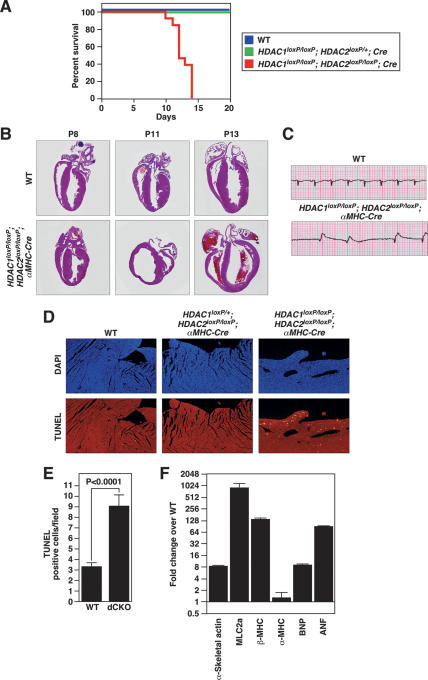
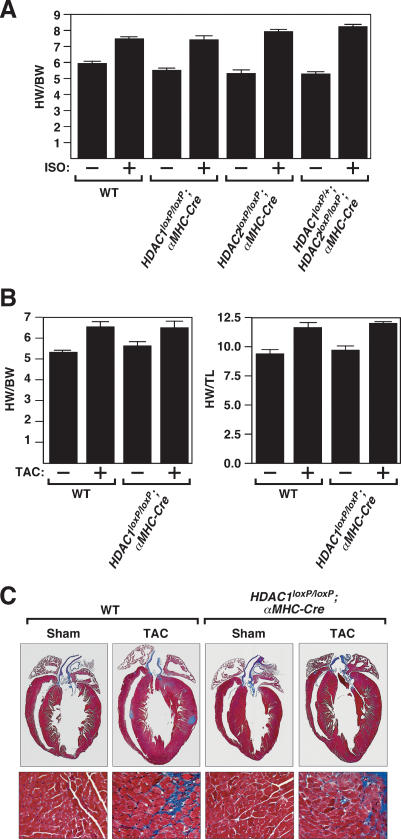
Histone deacetylases (HDACs) tighten chromatin structure and repress gene expression through the removal of acetyl groups from histone tails. The class I HDACs, HDAC1 and HDAC2, are expressed ubiquitously, but their potential roles in tissue-specific gene expression and organogenesis have not been defined. To explore the functions of HDAC1 and HDAC2 in vivo, we generated mice with conditional null alleles of both genes. Whereas global deletion of HDAC1 results in death by embryonic day 9.5, mice lacking HDAC2 survive until the perinatal period, when they succumb to a spectrum of cardiac defects, including obliteration of the lumen of the right ventricle, excessive hyperplasia and apoptosis of cardiomyocytes, and bradycardia. Cardiac-specific deletion of either HDAC1 or HDAC2 does not evoke a phenotype, whereas cardiac-specific deletion of both genes results in neonatal lethality, accompanied by cardiac arrhythmias, dilated cardiomyopathy, and up-regulation of genes encoding skeletal muscle-specific contractile proteins and calcium channels. Our results reveal cell-autonomous and non-cell-autonomous functions for HDAC1 and HDAC2 in the control of myocardial growth, morphogenesis, and contractility, which reflect partially redundant roles of these enzymes in tissue-specific transcriptional repression.
Figures


Similar articles
-
Comparison of Two Modern Survival Prediction Tools, SORG-MLA and METSSS, in Patients With Symptomatic Long-bone Metastases Who Underwent Local Treatment With Surgery Followed by Radiotherapy and With Radiotherapy Alone.Clin Orthop Relat Res. 2024 Dec 1;482(12):2193-2208. doi: 10.1097/CORR.0000000000003185. Epub 2024 Jul 23. Clin Orthop Relat Res. 2024. PMID: 39051924
-
Essential Nonredundant Function of the Catalytic Activity of Histone Deacetylase 2 in Mouse Development.Mol Cell Biol. 2015 Nov 23;36(3):462-74. doi: 10.1128/MCB.00639-15. Print 2016 Feb 1. Mol Cell Biol. 2015. PMID: 26598605 Free PMC article.
-
Falls prevention interventions for community-dwelling older adults: systematic review and meta-analysis of benefits, harms, and patient values and preferences.Syst Rev. 2024 Nov 26;13(1):289. doi: 10.1186/s13643-024-02681-3. Syst Rev. 2024. PMID: 39593159 Free PMC article.
-
Depressing time: Waiting, melancholia, and the psychoanalytic practice of care.In: Kirtsoglou E, Simpson B, editors. The Time of Anthropology: Studies of Contemporary Chronopolitics. Abingdon: Routledge; 2020. Chapter 5. In: Kirtsoglou E, Simpson B, editors. The Time of Anthropology: Studies of Contemporary Chronopolitics. Abingdon: Routledge; 2020. Chapter 5. PMID: 36137063 Free Books & Documents. Review.
-
Trends in Surgical and Nonsurgical Aesthetic Procedures: A 14-Year Analysis of the International Society of Aesthetic Plastic Surgery-ISAPS.Aesthetic Plast Surg. 2024 Oct;48(20):4217-4227. doi: 10.1007/s00266-024-04260-2. Epub 2024 Aug 5. Aesthetic Plast Surg. 2024. PMID: 39103642 Review.
Cited by
-
A conserved gammaherpesvirus protein kinase targets histone deacetylases 1 and 2 to facilitate viral replication in primary macrophages.J Virol. 2013 Jul;87(13):7314-25. doi: 10.1128/JVI.02713-12. Epub 2013 Apr 24. J Virol. 2013. PMID: 23616648 Free PMC article.
-
Targeting histone deacetylase in cardiac diseases.Front Physiol. 2024 Jun 24;15:1405569. doi: 10.3389/fphys.2024.1405569. eCollection 2024. Front Physiol. 2024. PMID: 38983721 Free PMC article. Review.
-
Epigenetics of the failing heart.Heart Fail Rev. 2015 Jul;20(4):435-59. doi: 10.1007/s10741-015-9483-x. Heart Fail Rev. 2015. PMID: 25847519 Review.
-
HDAC1 regulates pluripotency and lineage specific transcriptional networks in embryonic and trophoblast stem cells.Nucleic Acids Res. 2012 Apr;40(7):2925-39. doi: 10.1093/nar/gkr1151. Epub 2011 Dec 10. Nucleic Acids Res. 2012. PMID: 22156375 Free PMC article.
-
A recurrent de novo CTBP1 mutation is associated with developmental delay, hypotonia, ataxia, and tooth enamel defects.Neurogenetics. 2016 Jul;17(3):173-8. doi: 10.1007/s10048-016-0482-4. Epub 2016 Apr 19. Neurogenetics. 2016. PMID: 27094857
References
-
- Agah R., Frenkel P.A., French B.A., Michael L.H., Overbeek P.A., Schneider M.D., Frenkel P.A., French B.A., Michael L.H., Overbeek P.A., Schneider M.D., French B.A., Michael L.H., Overbeek P.A., Schneider M.D., Michael L.H., Overbeek P.A., Schneider M.D., Overbeek P.A., Schneider M.D., Schneider M.D. Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J. Clin. Invest. 1997;100:169–179. - PMC - PubMed
-
- Antos C.L., McKinsey T.A., Dreitz M., Hollingsworth L.M., Zhang C.L., Schreiber K., Rindt H., Gorczynski R.J., Olson E.N., McKinsey T.A., Dreitz M., Hollingsworth L.M., Zhang C.L., Schreiber K., Rindt H., Gorczynski R.J., Olson E.N., Dreitz M., Hollingsworth L.M., Zhang C.L., Schreiber K., Rindt H., Gorczynski R.J., Olson E.N., Hollingsworth L.M., Zhang C.L., Schreiber K., Rindt H., Gorczynski R.J., Olson E.N., Zhang C.L., Schreiber K., Rindt H., Gorczynski R.J., Olson E.N., Schreiber K., Rindt H., Gorczynski R.J., Olson E.N., Rindt H., Gorczynski R.J., Olson E.N., Gorczynski R.J., Olson E.N., Olson E.N. Dose-dependent blockade to cardiomyocyte hypertrophy by histone deacetylase inhibitors. J. Biol. Chem. 2003;278:28930–28937. - PubMed
-
- Bodor G.S., Oakeley A.E., Allen P.D., Crimmins D.L., Ladenson J.H., Anderson P.A., Oakeley A.E., Allen P.D., Crimmins D.L., Ladenson J.H., Anderson P.A., Allen P.D., Crimmins D.L., Ladenson J.H., Anderson P.A., Crimmins D.L., Ladenson J.H., Anderson P.A., Ladenson J.H., Anderson P.A., Anderson P.A. Troponin I phosphorylation in the normal and failing adult human heart. Circulation. 1997;96:1495–1500. - PubMed
-
- Bolden J.E., Peart M.J., Johnstone R.W., Peart M.J., Johnstone R.W., Johnstone R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006;5:769–784. - PubMed
-
- Davis C.A., Haberland M., Arnold M.A., Sutherland L.B., McDonald O.G., Richardson J.A., Childs G., Harris S., Owens G.K., Olson E.N., Haberland M., Arnold M.A., Sutherland L.B., McDonald O.G., Richardson J.A., Childs G., Harris S., Owens G.K., Olson E.N., Arnold M.A., Sutherland L.B., McDonald O.G., Richardson J.A., Childs G., Harris S., Owens G.K., Olson E.N., Sutherland L.B., McDonald O.G., Richardson J.A., Childs G., Harris S., Owens G.K., Olson E.N., McDonald O.G., Richardson J.A., Childs G., Harris S., Owens G.K., Olson E.N., Richardson J.A., Childs G., Harris S., Owens G.K., Olson E.N., Childs G., Harris S., Owens G.K., Olson E.N., Harris S., Owens G.K., Olson E.N., Owens G.K., Olson E.N., Olson E.N. PRISM/PRDM6, a transcriptional repressor that promotes the proliferative gene program in smooth muscle cells. Mol. Cell. Biol. 2006;26:2626–2636. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous