

Frontotemporal lobar degeneration: current concepts in the light of recent advances
- PMID: 17493044
- PMCID: PMC8095552
- DOI: 10.1111/j.1750-3639.2007.00055.x
Frontotemporal lobar degeneration: current concepts in the light of recent advances
Abstract
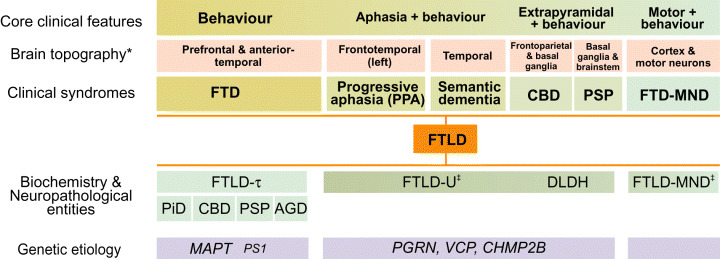
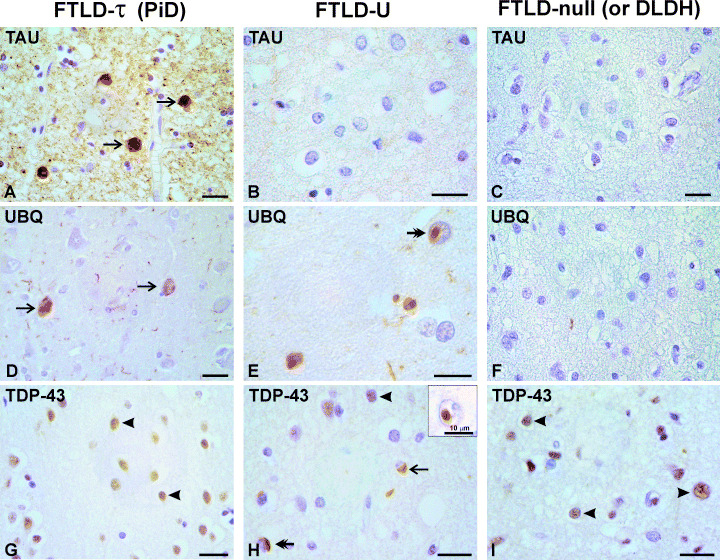
Work done over the past decade has led to a molecular understanding of frontotemporal lobar degeneration (FTLD), a deadly disease that afflicts patients in mid-life. It is a common cause of dementia, second only to Alzheimer's disease in the population below 65 years of age. Neuroanatomical and neurobiological substrates have been identified for the three major subtypes of FTLD and these discoveries have broadened the FTLD spectrum to include amyotrophic lateral sclerosis (ALS). Mutations in MAPT were found to cause frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17), a familial disorder with filamentous tau inclusions in nerve cells and glial cells. FTDP-17 can result in clinical syndromes that closely resemble progressive supranuclear palsy, corticobasal degeneration and Pick's disease. More recently, mutations in three genes (VCP, CHMP2B and PGRN) have been found to cause FTLD with ubiquitin-positive, tau-negative neuronal inclusions (FTLD-U). They explain a large proportion of inherited FTLD-U. It remains to be seen whether dementia lacking distinctive histopathology (DLDH) constitutes a third disease category, as many of these cases are now being reclassified as FTLD-U. Recently, TAR DNA-binding protein-43 (TDP-43) has been identified as a key protein of the ubiquitin inclusions of FTLD-U and ALS. Thus, for familial forms of FTLD and related disorders, we now know the primary etiologies and accumulating proteins. These findings are pivotal for dissecting the pathways by which different etiologies lead to the varied clinicopathological presentations of FTLD.
Figures
Similar articles
-
Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration.Acta Neuropathol. 2007 Jul;114(1):5-22. doi: 10.1007/s00401-007-0237-2. Epub 2007 Jun 20. Acta Neuropathol. 2007. PMID: 17579875 Free PMC article.
-
Retiring the term FTDP-17 as MAPT mutations are genetic forms of sporadic frontotemporal tauopathies.Brain. 2018 Feb 1;141(2):521-534. doi: 10.1093/brain/awx328. Brain. 2018. PMID: 29253099 Free PMC article.
-
Frontotemporal lobar degeneration--tau as a pied piper?Neurogenetics. 2002 Oct;4(2):63-75. doi: 10.1007/s10048-002-0140-x. Neurogenetics. 2002. PMID: 12481984 Review.
-
Clinicopathological characterization of Pick's disease versus frontotemporal lobar degeneration with ubiquitin/TDP-43-positive inclusions.Acta Neuropathol. 2009 Apr;117(4):429-44. doi: 10.1007/s00401-009-0493-4. Epub 2009 Feb 5. Acta Neuropathol. 2009. PMID: 19194716
-
Pathology and genetics of frontotemporal lobar degeneration: an update.Clin Neuropathol. 2007 Jul-Aug;26(4):143-56. doi: 10.5414/npp26143. Clin Neuropathol. 2007. PMID: 17702495 Review.
Cited by
-
Progranulin and TDP-43: mechanistic links and future directions.J Mol Neurosci. 2011 Nov;45(3):561-73. doi: 10.1007/s12031-011-9625-0. Epub 2011 Aug 24. J Mol Neurosci. 2011. PMID: 21863317 Free PMC article. Review.
-
Multiplex SILAC analysis of a cellular TDP-43 proteinopathy model reveals protein inclusions associated with SUMOylation and diverse polyubiquitin chains.Mol Cell Proteomics. 2010 Apr;9(4):705-18. doi: 10.1074/mcp.M800390-MCP200. Epub 2010 Jan 4. Mol Cell Proteomics. 2010. PMID: 20047951 Free PMC article.
-
TDP-43: a novel neurodegenerative proteinopathy.Curr Opin Neurobiol. 2007 Oct;17(5):548-55. doi: 10.1016/j.conb.2007.08.005. Epub 2007 Oct 23. Curr Opin Neurobiol. 2007. PMID: 17936612 Free PMC article.
-
Knockdown of transactive response DNA-binding protein (TDP-43) downregulates histone deacetylase 6.EMBO J. 2010 Jan 6;29(1):209-21. doi: 10.1038/emboj.2009.324. Epub 2009 Nov 12. EMBO J. 2010. PMID: 19910924 Free PMC article.
-
The novel MAPT mutation K298E: mechanisms of mutant tau toxicity, brain pathology and tau expression in induced fibroblast-derived neurons.Acta Neuropathol. 2014 Feb;127(2):283-95. doi: 10.1007/s00401-013-1219-1. Epub 2013 Nov 30. Acta Neuropathol. 2014. PMID: 24292008 Free PMC article.
References
-
- Amtul Z, Lewis PA, Piper S, Crook R, Baker M, Findlay K, Singleton A, Hogg M, Younkin L, Younkin SG, Hardy J, Hutton M, Boeve BF, Tang‐Wai D, Golde TE (2002) A presenilin 1 mutation associated with familial frontotemporal dementia inhibits gamma‐secretase cleavage of APP and notch. Neurobiol Dis 9:269–273. - PubMed
-
- Arai T, Nonaka T, Hasegawa M, Akiyama H, Yoshida M, Hashizume Y, Tsuchiya K, Oda T, Ikeda K (2003) Neuronal and glial inclusions in frontotemporal dementia with or without motor neuron disease are immunopositive for p62. Neurosci Lett 342:41–44. - PubMed
-
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T (2006) TDP‐43 is a component of ubiquitin‐positive tau‐negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351:602–611. - PubMed
-
- Babst M, Katzmann DJ, Estepa‐Sabal EJ, Meerloo T, Emr SD (2002) Escrt‐III: an endosome‐associated heterooligomeric protein complex required for mvb sorting. Dev Cell 3:271–282. - PubMed
-
- Baker M, Litvan I, Houlden H, Adamson J, Dickson D, Perez‐tur J, Hardy J, Lynch T, Bigio E, Hutton M (1999) Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum Mol Genet 8:711–715. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
