

Neurotrophic and neuroprotective actions of estrogen: basic mechanisms and clinical implications
- PMID: 17379265
- PMCID: PMC2048656
- DOI: 10.1016/j.steroids.2007.02.003
Neurotrophic and neuroprotective actions of estrogen: basic mechanisms and clinical implications
Abstract
Estrogen is an important hormone signal that regulates multiple tissues and functions in the body. This review focuses on the neurotrophic and neuroprotective actions of estrogen in the brain, with particular emphasis on estrogen actions in the hippocampus, cerebral cortex and striatum. Sex differences in the risk, onset and severity of neurodegenerative disease such as Alzheimer's disease, Parkinson's disease and stroke are well known, and the potential role of estrogen as a neuroprotective factor is discussed in this context. The review assimilates a complex literature that spans research in humans, non-human primates and rodent animal models and attempts to contrast and compare the findings across species where possible. Current controversies regarding the Women's Health Initiative (WHI) study, its ramifications, concerns and the new studies needed to address these concerns are also addressed. Signaling mechanisms underlying estrogen-induced neuroprotection and synaptic plasticity are reviewed, including the important concepts of genomic versus nongenomic mechanisms, types of estrogen receptor involved and their subcellular targeting, and implicated downstream signaling pathways and mediators. Finally, a multicellular mode of estrogen action in the regulation of neuronal survival and neurotrophism is discussed, as are potential future directions for the field.
Figures
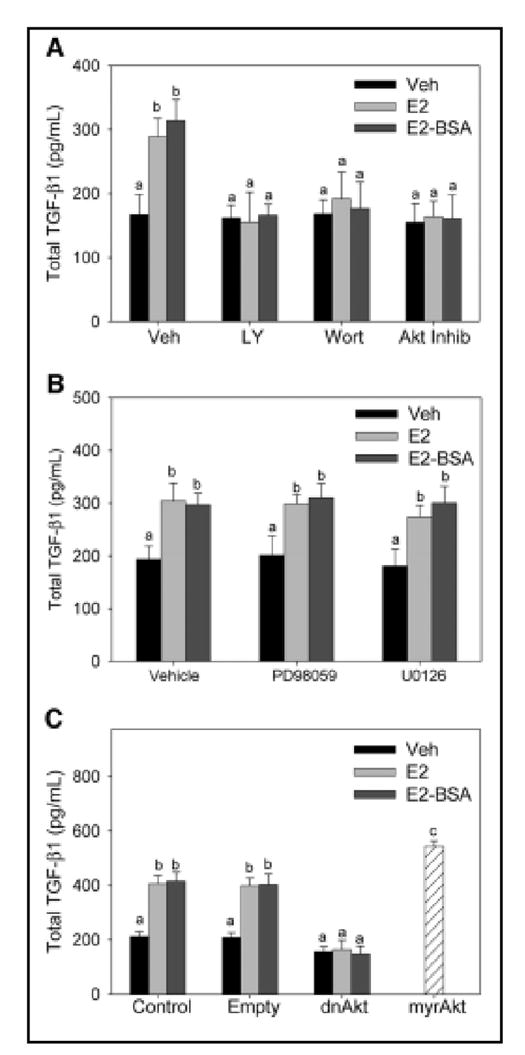
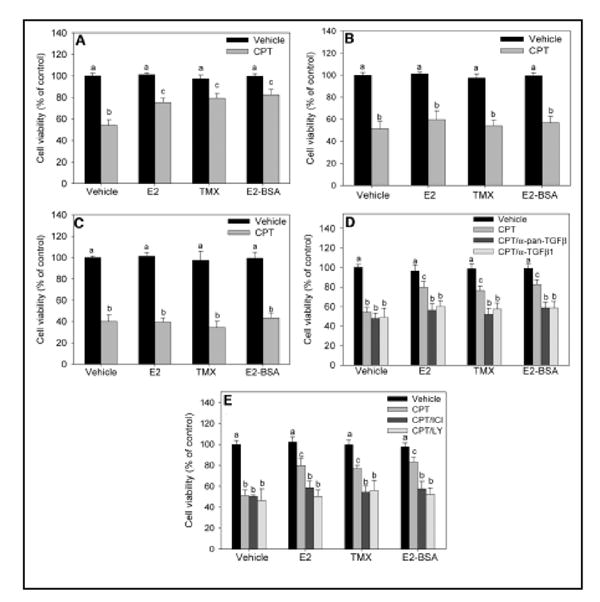
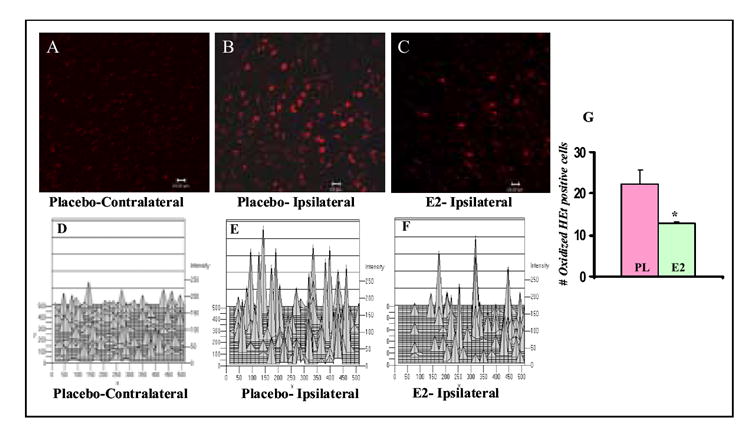
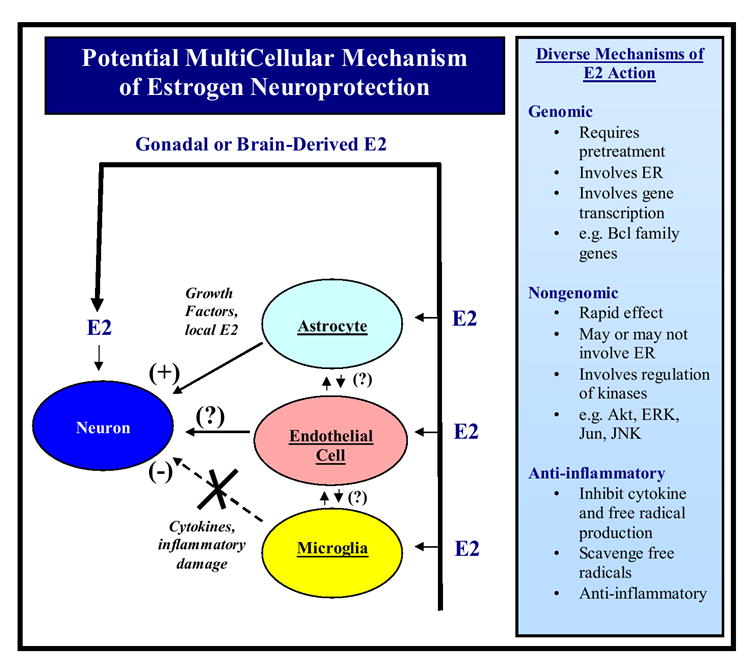
Similar articles
-
Oestrogen as a neuroprotective hormone.Nat Rev Neurosci. 2002 Jun;3(6):433-42. doi: 10.1038/nrn846. Nat Rev Neurosci. 2002. PMID: 12042878 Review.
-
Neuroprotective effects of estrogens: potential mechanisms of action.Int J Dev Neurosci. 2000 Jul-Aug;18(4-5):347-58. doi: 10.1016/s0736-5748(00)00017-4. Int J Dev Neurosci. 2000. PMID: 10817919 Review.
-
Select estrogens within the complex formulation of conjugated equine estrogens (Premarin) are protective against neurodegenerative insults: implications for a composition of estrogen therapy to promote neuronal function and prevent Alzheimer's disease.BMC Neurosci. 2006 Mar 13;7:24. doi: 10.1186/1471-2202-7-24. BMC Neurosci. 2006. PMID: 16533397 Free PMC article.
-
Effects of estrogen in the brain: is it a neuroprotective agent in Alzheimer's disease?Curr Aging Sci. 2010 Jul;3(2):113-26. doi: 10.2174/1874609811003020113. Curr Aging Sci. 2010. PMID: 20167003 Review.
-
Neuroprotective actions of sex steroids in Parkinson's disease.Front Neuroendocrinol. 2009 Jul;30(2):142-57. doi: 10.1016/j.yfrne.2009.04.014. Epub 2009 May 3. Front Neuroendocrinol. 2009. PMID: 19410597 Review.
Cited by
-
Longitudinal monitoring of the mouse brain reveals heterogenous network trajectories during aging.Commun Biol. 2024 Feb 20;7(1):210. doi: 10.1038/s42003-024-05873-8. Commun Biol. 2024. PMID: 38378942 Free PMC article.
-
Influences of hormone replacement therapy on olfactory and cognitive function in postmenopausal women.Neurobiol Aging. 2015 Jun;36(6):2053-9. doi: 10.1016/j.neurobiolaging.2015.02.028. Epub 2015 Mar 10. Neurobiol Aging. 2015. PMID: 25850354 Free PMC article.
-
Estradiol enhanced neuronal plasticity and ameliorated astrogliosis in human iPSC-derived neural models.Regen Ther. 2024 Jan 12;25:250-263. doi: 10.1016/j.reth.2023.12.018. eCollection 2024 Mar. Regen Ther. 2024. PMID: 38293585 Free PMC article.
-
Synaptic estrogen receptor-alpha levels in prefrontal cortex in female rhesus monkeys and their correlation with cognitive performance.J Neurosci. 2010 Sep 22;30(38):12770-6. doi: 10.1523/JNEUROSCI.3192-10.2010. J Neurosci. 2010. PMID: 20861381 Free PMC article.
-
Oophorectomy Reduces Estradiol Levels and Long-Term Spontaneous Neurovascular Recovery in a Female Rat Model of Focal Ischemic Stroke.Front Mol Neurosci. 2018 Sep 13;11:338. doi: 10.3389/fnmol.2018.00338. eCollection 2018. Front Mol Neurosci. 2018. PMID: 30271324 Free PMC article.
References
-
- Petersen SL, Ottem EN, Carpenter CD. Direct and indirect regulation of gonadotropin-releasing hormone neurons by estrogen. Biol Reprod. 2003;69:17771–1778. - PubMed
-
- Kelly MJ, Qiu J, Ronnekleiv OK. Estrogen signaling in the hypothalamus. Vitam Horm. 2005;71:123–145. - PubMed
-
- Brann DW, Mills TM, Mahesh VB. Female reproduction the ovulatory cycle . In: Witorsch RJ, editor. Reproductive Toxicology. 2. Raven Press; 1995. pp. 23–44.
-
- Hewitt SC, Korach KS. Oestrogen receptor knockout mice: roles for oestrogen receptors alpha and beta in reproductive tissues. Reproduction. 2003;125:143–149. - PubMed
-
- Gore AC. Gonadotropin-releasing hormone neurons, NMDA receptors, and their regulation by steroid hormones across the reproductive life cycle. Brain Res Brain Res Rev. 2001;37:235–248. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
