

Notch signaling is necessary for epithelial growth arrest by TGF-beta
- PMID: 17325209
- PMCID: PMC2064026
- DOI: 10.1083/jcb.200612129
Notch signaling is necessary for epithelial growth arrest by TGF-beta
Abstract
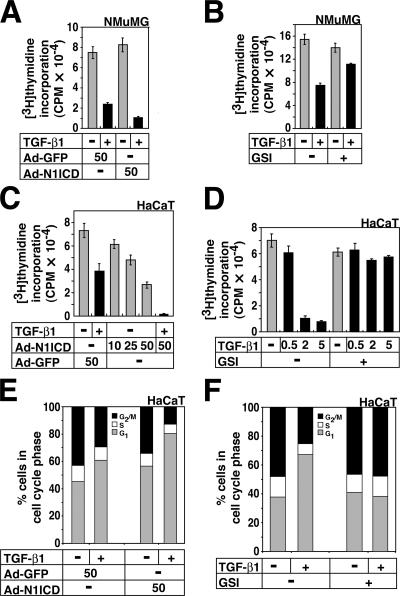
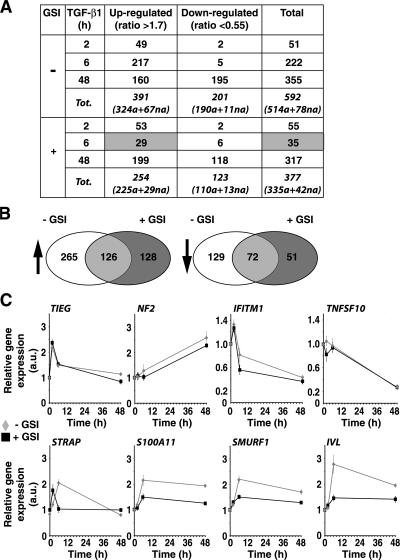
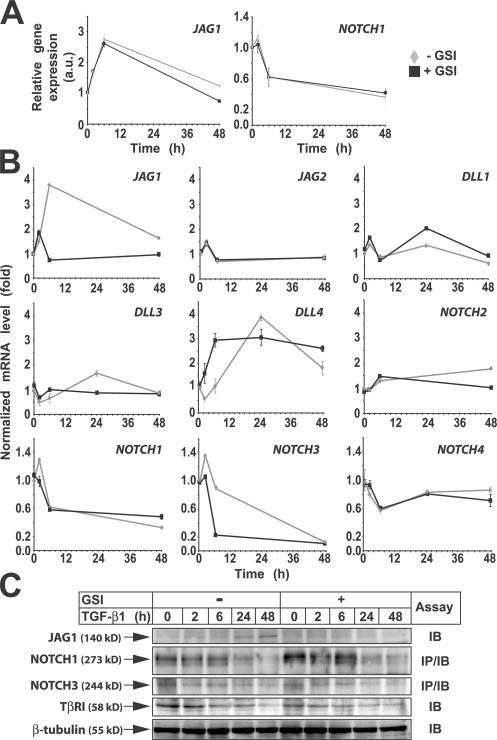
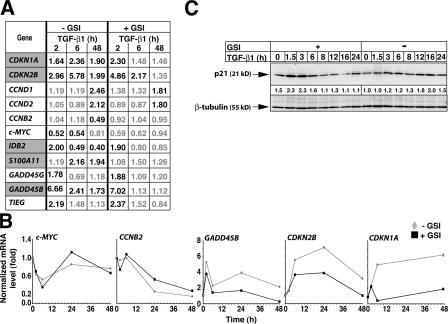
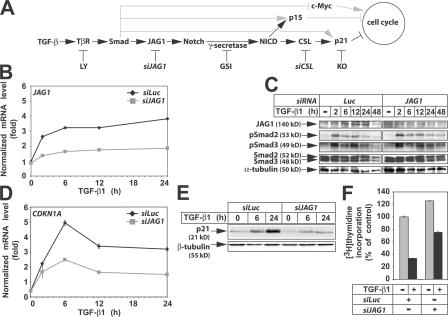
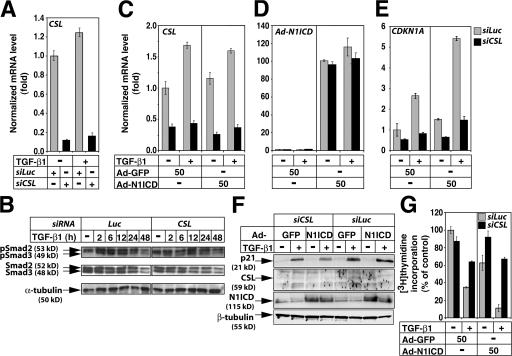
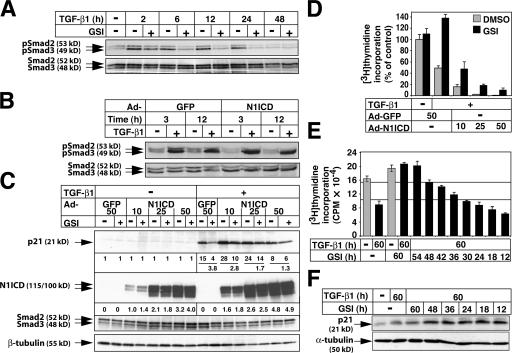
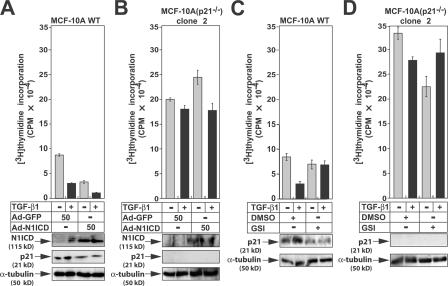
Transforming growth factor beta (TGF-beta) and Notch act as tumor suppressors by inhibiting epithelial cell proliferation. TGF-beta additionally promotes tumor invasiveness and metastasis, whereas Notch supports oncogenic growth. We demonstrate that TGF-beta and ectopic Notch1 receptor cooperatively arrest epithelial growth, whereas endogenous Notch signaling was found to be required for TGF-beta to elicit cytostasis. Transcriptomic analysis after blocking endogenous Notch signaling uncovered several genes, including Notch pathway components and cell cycle and apoptosis factors, whose regulation by TGF-beta requires an active Notch pathway. A prominent gene coregulated by the two pathways is the cell cycle inhibitor p21. Both transcriptional induction of the Notch ligand Jagged1 by TGF-beta and endogenous levels of the Notch effector CSL contribute to p21 induction and epithelial cytostasis. Cooperative inhibition of cell proliferation by TGF-beta and Notch is lost in human mammary cells in which the p21 gene has been knocked out. We establish an intimate involvement of Notch signaling in the epithelial cytostatic response to TGF-beta.
Figures
Similar articles
-
Blockade of Jagged/Notch pathway abrogates transforming growth factor β2-induced epithelial-mesenchymal transition in human retinal pigment epithelium cells.Curr Mol Med. 2014 May;14(4):523-34. doi: 10.2174/1566524014666140331230411. Curr Mol Med. 2014. PMID: 24694299 Review.
-
Notch1 signaling and regulatory T cell function.J Immunol. 2008 Mar 1;180(5):2796-804. doi: 10.4049/jimmunol.180.5.2796. J Immunol. 2008. PMID: 18292500
-
A role for notch signaling in human corneal epithelial cell differentiation and proliferation.Invest Ophthalmol Vis Sci. 2007 Aug;48(8):3576-85. doi: 10.1167/iovs.06-1373. Invest Ophthalmol Vis Sci. 2007. PMID: 17652726
-
Notch signaling mediates TGF-β1-induced epithelial-mesenchymal transition through the induction of Snai1.Int J Biochem Cell Biol. 2012 May;44(5):776-89. doi: 10.1016/j.biocel.2012.01.021. Epub 2012 Feb 7. Int J Biochem Cell Biol. 2012. PMID: 22330899
-
Mechanisms of TGF-beta signaling in regulation of cell growth and differentiation.Immunol Lett. 2002 Jun 3;82(1-2):85-91. doi: 10.1016/s0165-2478(02)00023-8. Immunol Lett. 2002. PMID: 12008039 Review.
Cited by
-
Different levels of Notch signaling regulate quiescence, renewal and differentiation in pancreatic endocrine progenitors.Development. 2012 May;139(9):1557-67. doi: 10.1242/dev.076000. Development. 2012. PMID: 22492351 Free PMC article.
-
Notch signalling in solid tumours: a little bit of everything but not all the time.Nat Rev Cancer. 2011 May;11(5):338-51. doi: 10.1038/nrc3035. Epub 2011 Apr 14. Nat Rev Cancer. 2011. PMID: 21508972 Review.
-
The role of Notch signaling pathway in epithelial-mesenchymal transition (EMT) during development and tumor aggressiveness.Curr Drug Targets. 2010 Jun;11(6):745-51. doi: 10.2174/138945010791170860. Curr Drug Targets. 2010. PMID: 20041844 Free PMC article. Review.
-
Role of Notch and its oncogenic signaling crosstalk in breast cancer.Biochim Biophys Acta. 2011 Apr;1815(2):197-213. doi: 10.1016/j.bbcan.2010.12.002. Epub 2010 Dec 28. Biochim Biophys Acta. 2011. PMID: 21193018 Free PMC article. Review.
-
Can Natural Products Targeting EMT Serve as the Future Anticancer Therapeutics?Molecules. 2022 Nov 8;27(22):7668. doi: 10.3390/molecules27227668. Molecules. 2022. PMID: 36431766 Free PMC article. Review.
References
-
- Bachman, K.E., B.G. Blair, K. Brenner, A. Bardelli, S. Arena, S. Zhou, J. Hicks, A.M. De Marzo, P. Argani, and B.H. Park. 2004. p21WAF1/CIP1 mediates the growth response to TGF-β in human epithelial cells. Cancer Biol. Ther. 3:221–225. - PubMed
-
- Brunkan, A.L., and A.M. Goate. 2005. Presenilin function and γ-secretase activity. J. Neurochem. 93:769–792. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
