

Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta
- PMID: 17277775
- PMCID: PMC7510175
- DOI: 10.1038/ng1968
Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta
Erratum in
- Nat Genet. 2008 Jul;40(7):927
Abstract
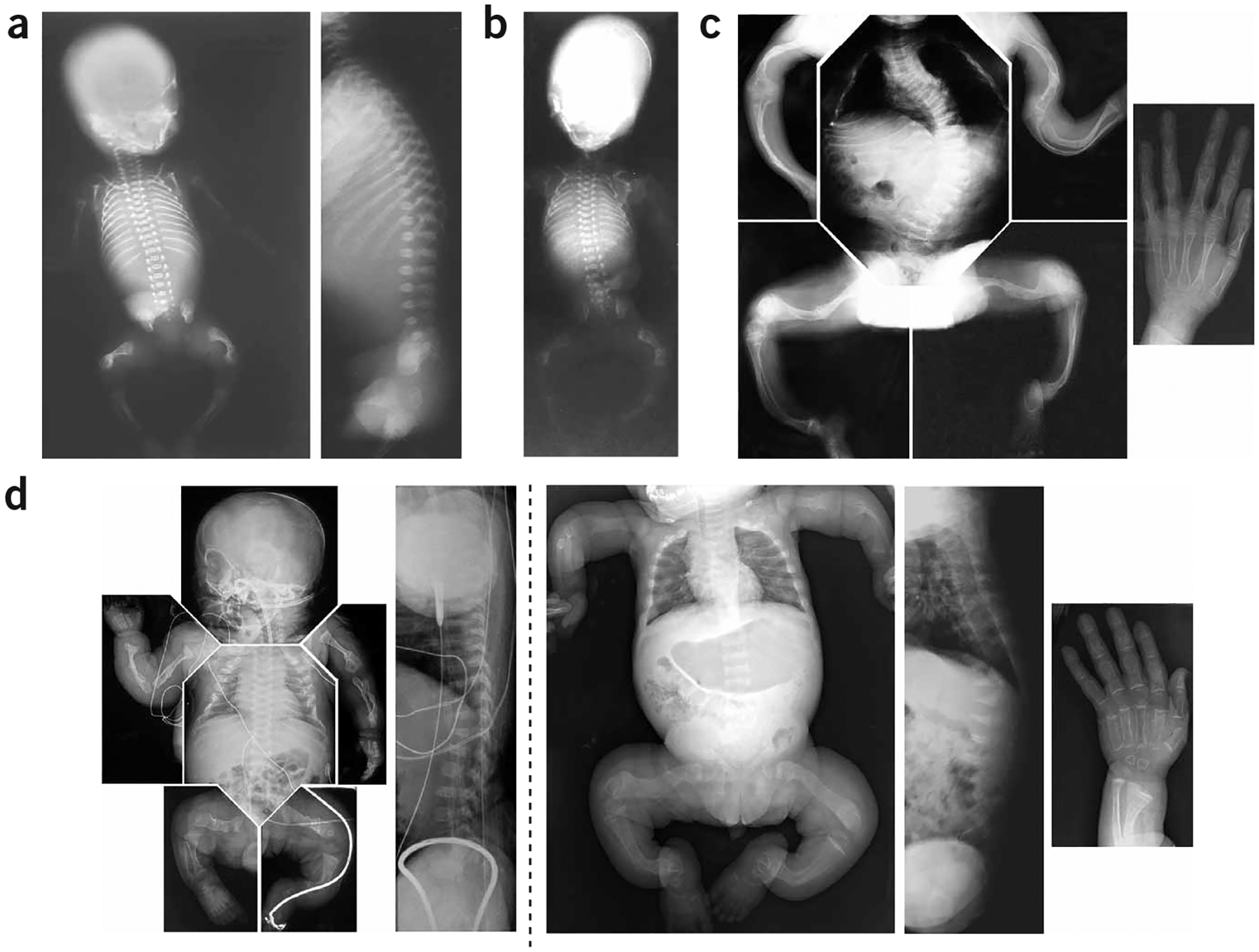
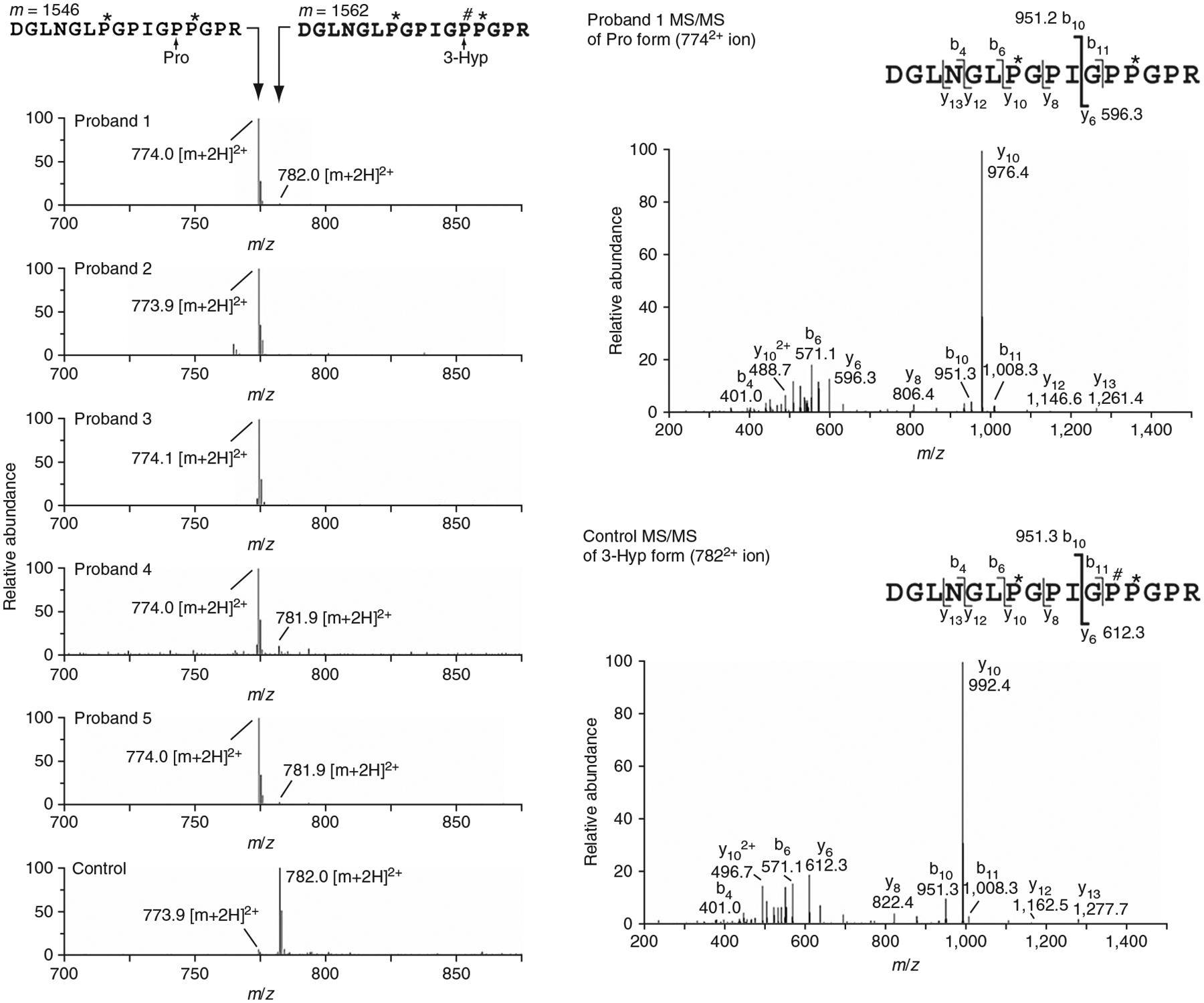
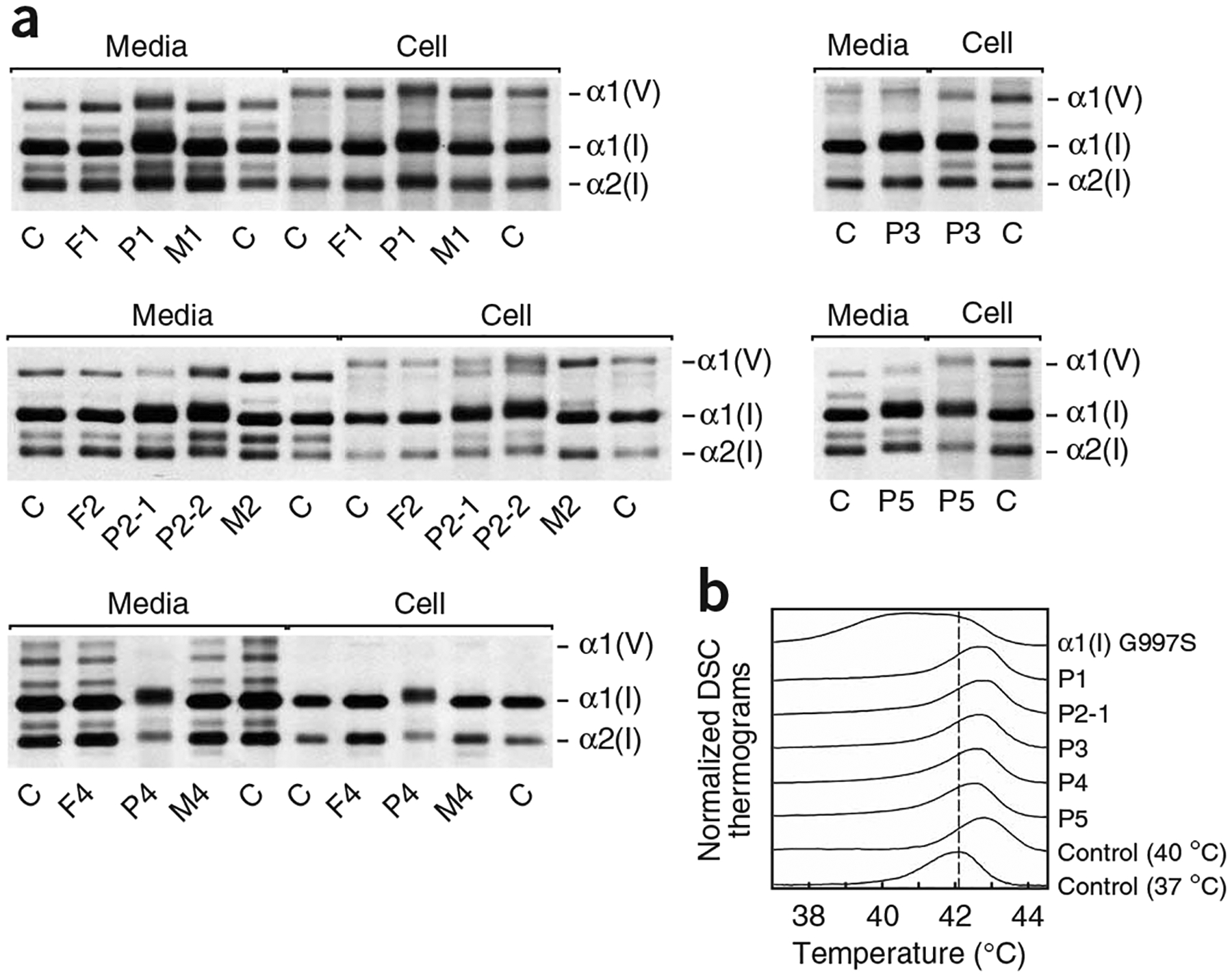
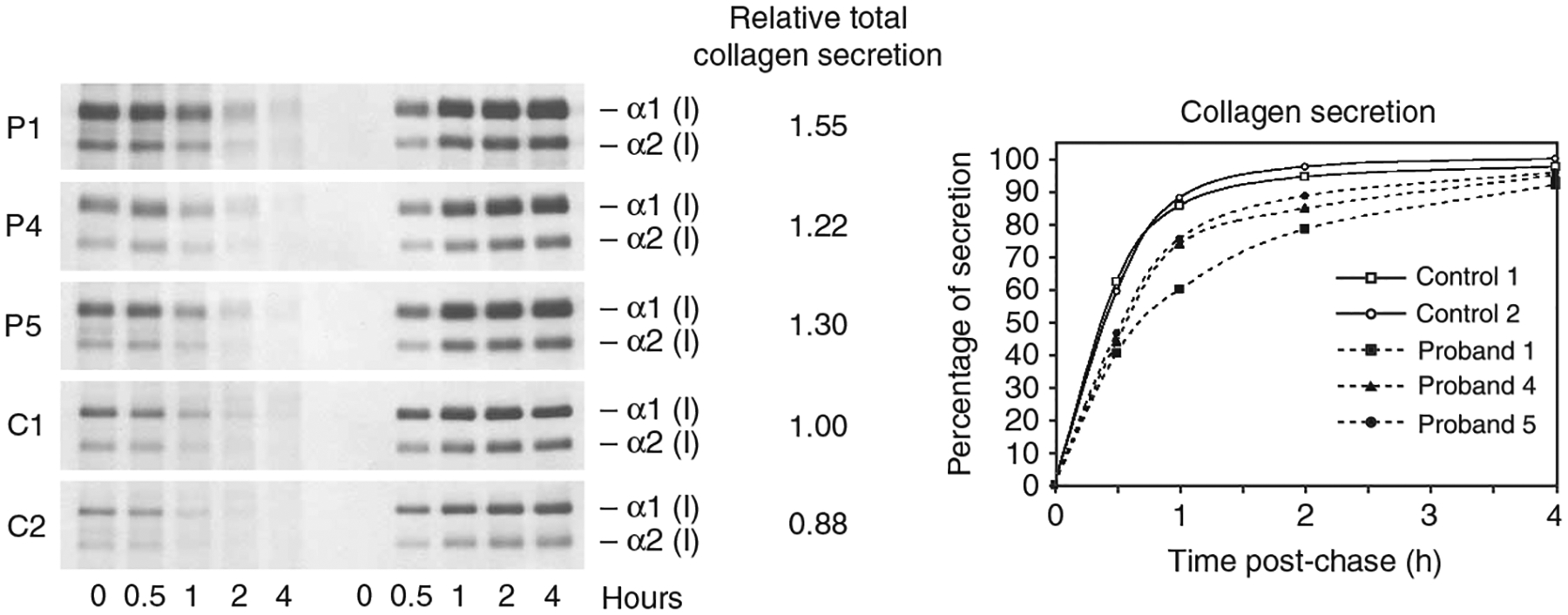
A recessive form of severe osteogenesis imperfecta that is not caused by mutations in type I collagen has long been suspected. Mutations in human CRTAP (cartilage-associated protein) causing recessive bone disease have been reported. CRTAP forms a complex with cyclophilin B and prolyl 3-hydroxylase 1, which is encoded by LEPRE1 and hydroxylates one residue in type I collagen, alpha1(I)Pro986. We present the first five cases of a new recessive bone disorder resulting from null LEPRE1 alleles; its phenotype overlaps with lethal/severe osteogenesis imperfecta but has distinctive features. Furthermore, a mutant allele from West Africa, also found in African Americans, occurs in four of five cases. All proband LEPRE1 mutations led to premature termination codons and minimal mRNA and protein. Proband collagen had minimal 3-hydroxylation of alpha1(I)Pro986 but excess lysyl hydroxylation and glycosylation along the collagen helix. Proband collagen secretion was moderately delayed, but total collagen secretion was increased. Prolyl 3-hydroxylase 1 is therefore crucial for bone development and collagen helix formation.
Conflict of interest statement
COMPETING INTERESTS STATEMENT
The authors declare that they have no competing financial interests.
Figures

Similar articles
-
Null mutations in LEPRE1 and CRTAP cause severe recessive osteogenesis imperfecta.Cell Tissue Res. 2010 Jan;339(1):59-70. doi: 10.1007/s00441-009-0872-0. Epub 2009 Oct 28. Cell Tissue Res. 2010. PMID: 19862557 Free PMC article. Review.
-
Recessive osteogenesis imperfecta caused by LEPRE1 mutations: clinical documentation and identification of the splice form responsible for prolyl 3-hydroxylation.J Med Genet. 2009 Apr;46(4):233-41. doi: 10.1136/jmg.2008.062729. Epub 2008 Dec 16. J Med Genet. 2009. PMID: 19088120
-
Deficiency of CRTAP in non-lethal recessive osteogenesis imperfecta reduces collagen deposition into matrix.Clin Genet. 2012 Nov;82(5):453-9. doi: 10.1111/j.1399-0004.2011.01794.x. Epub 2011 Oct 19. Clin Genet. 2012. PMID: 21955071 Free PMC article.
-
Mutations in PPIB (cyclophilin B) delay type I procollagen chain association and result in perinatal lethal to moderate osteogenesis imperfecta phenotypes.Hum Mol Genet. 2011 Apr 15;20(8):1595-609. doi: 10.1093/hmg/ddr037. Epub 2011 Jan 31. Hum Mol Genet. 2011. PMID: 21282188 Free PMC article.
-
Components of the collagen prolyl 3-hydroxylation complex are crucial for normal bone development.Cell Cycle. 2007 Jul 15;6(14):1675-81. doi: 10.4161/cc.6.14.4474. Epub 2007 May 18. Cell Cycle. 2007. PMID: 17630507 Review.
Cited by
-
Update on the Genetics of Osteogenesis Imperfecta.Calcif Tissue Int. 2024 Dec;115(6):891-914. doi: 10.1007/s00223-024-01266-5. Epub 2024 Aug 11. Calcif Tissue Int. 2024. PMID: 39127989 Free PMC article. Review.
-
A Dyadic Nosology for Osteogenesis Imperfecta and Bone Fragility Syndromes 2024.Calcif Tissue Int. 2024 Dec;115(6):873-890. doi: 10.1007/s00223-024-01248-7. Epub 2024 Jun 28. Calcif Tissue Int. 2024. PMID: 38942908 Free PMC article. Review.
-
Identification of domains and amino acids essential to the collagen galactosyltransferase activity of GLT25D1.PLoS One. 2011;6(12):e29390. doi: 10.1371/journal.pone.0029390. Epub 2011 Dec 21. PLoS One. 2011. PMID: 22216269 Free PMC article.
-
Signalling mediated by the endoplasmic reticulum stress transducer OASIS is involved in bone formation.Nat Cell Biol. 2009 Oct;11(10):1205-11. doi: 10.1038/ncb1963. Epub 2009 Sep 20. Nat Cell Biol. 2009. PMID: 19767743
-
Altered Sox9 and FGF signaling gene expression in Aga2 OI mice negatively affects linear growth.JCI Insight. 2023 Nov 8;8(21):e171984. doi: 10.1172/jci.insight.171984. JCI Insight. 2023. PMID: 37796615 Free PMC article.
References
-
- Byers PH & Cole WG Osteogenesis imperfecta in Connective Tissue and Its Heritable Disorders (eds. Royce PM & Steinmann B) 385–430 (Wiley-Liss, Inc., New York, 2002).
-
- Marini JC Osteogenesis imperfecta in Nelson Textbook of Pediatrics 17th ed (eds. Behrman RE, Kliegman RM & Jenson HB) 2336–2338 (Saunders, Philadelphia, 2004).
-
- Aitchison K, Ogilvie D, Honeyman M, Thompson E & Sykes B Homozygous osteogenesis imperfecta unlinked to collagen I genes. Hum. Genet 78, 233–236 (1988). - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
