

Neuropilin-1 is a direct target of the transcription factor E2F1 during cerebral ischemia-induced neuronal death in vivo
- PMID: 17178835
- PMCID: PMC1820462
- DOI: 10.1128/MCB.01760-06
Neuropilin-1 is a direct target of the transcription factor E2F1 during cerebral ischemia-induced neuronal death in vivo
Abstract
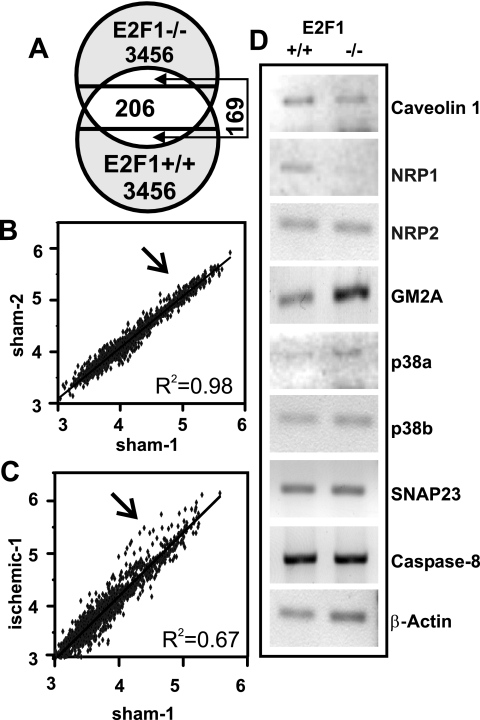
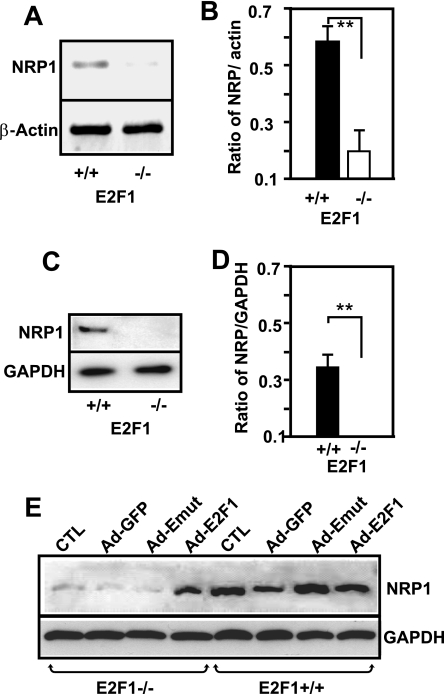
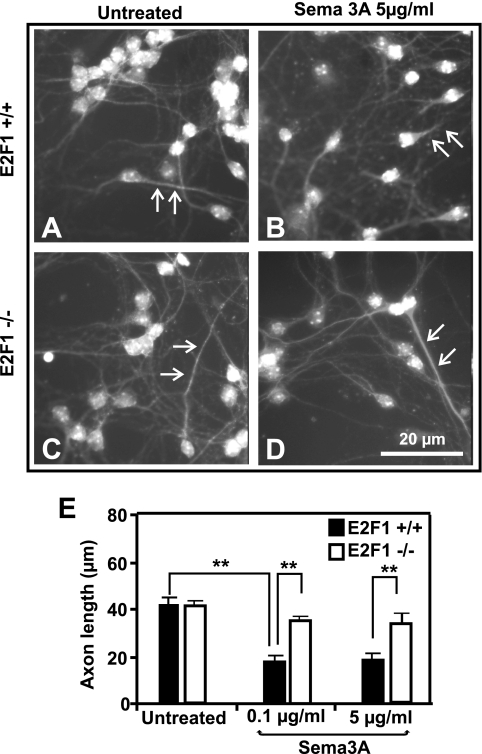
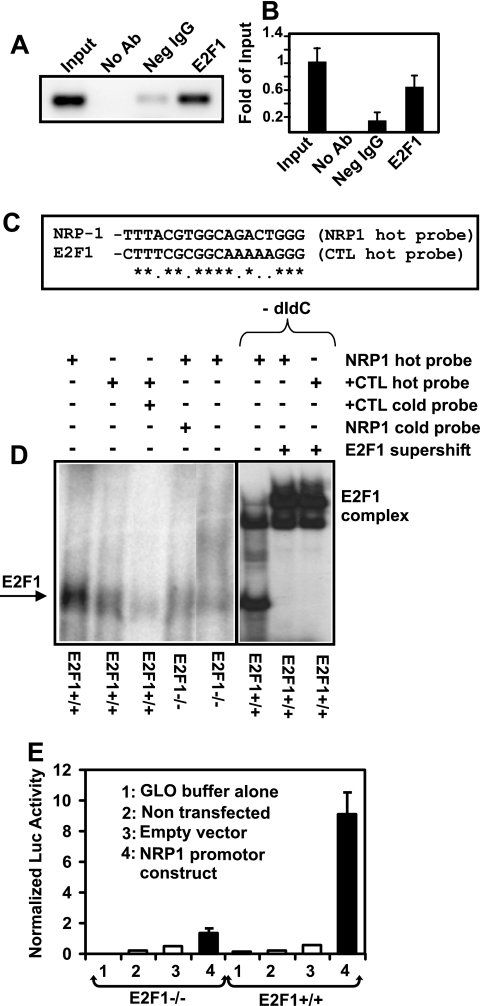
The nuclear transcription factor E2F1 plays an important role in modulating neuronal death in response to excitotoxicity and cerebral ischemia. Here, by comparing gene expression in brain cortices from E2F1(+/+) and E2F1(-/-) mice using a custom high-density DNA microarray, we identified a group of putative E2F1 target genes that might be responsible for ischemia-induced E2F1-dependent neuronal death. Neuropilin 1 (NRP-1), a receptor for semaphorin 3A-mediated axon growth cone collapse and retraction, was confirmed to be a direct target of E2F1 based on (i) the fact that the NRP-1 promoter sequence contains an E2F1 binding site, (ii) reactivation of NRP-1 expression in E2F1(-/-) neurons when the E2F1 gene was replaced, (iii) activation of the NRP-1 promoter by E2F1 in a luciferase reporter assay, (iv) electrophoretic mobility gel shift analysis confirmation of the presence of an E2F binding sequence in the NRP-1 promoter, and (v) the fact that a chromatin immunoprecipitation assay showed that E2F1 binds directly to the endogenous NRP-1 promoter. Interestingly, the temporal induction in cerebral ischemia-induced E2F1 binding to the NRP-1 promoter correlated with the temporal-induction profile of NRP-1 mRNA, confirming that E2F1 positively regulates NRP-1 during cerebral ischemia. Functional analysis also showed that NRP-1 receptor expression was extremely low in E2F1(-/-) neurons, which led to the diminished response to semaphorin 3A-induced axonal shortening and neuronal death. An NRP-1 selective peptide inhibitor provided neuroprotection against oxygen-glucose deprivation. Taken together, these findings support a model in which E2F1 targets NRP-1 to modulate axonal damage and neuronal death in response to cerebral ischemia.
Figures
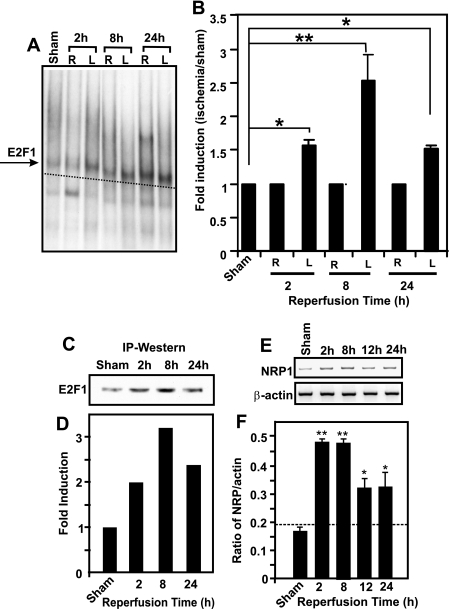
 and indicate statistical significance at P < 0.05 and P < 0.01, respectively, by one-way ANOVA with a post hoc Tukey's test for significant groups. The error bars indicate standard deviations.
and indicate statistical significance at P < 0.05 and P < 0.01, respectively, by one-way ANOVA with a post hoc Tukey's test for significant groups. The error bars indicate standard deviations.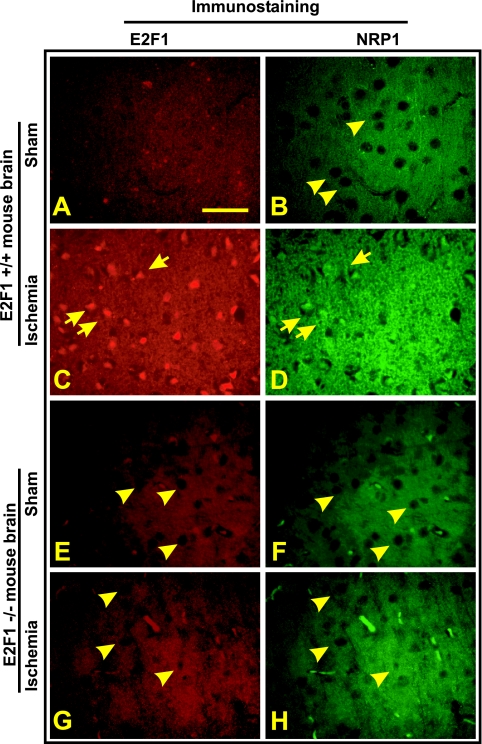
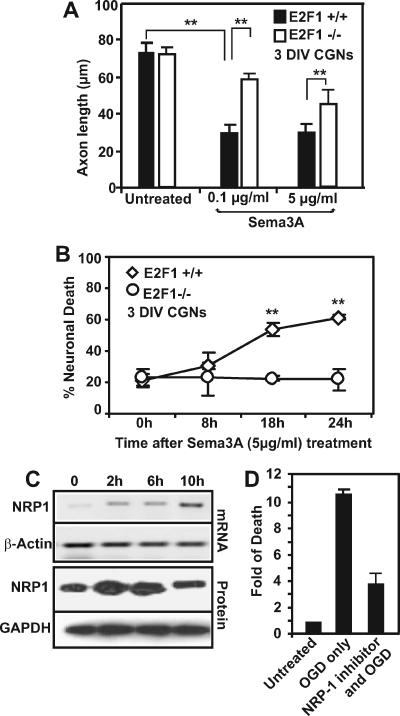 , P < 0.01 by ANOVA and post hoc Tukey's test. (C) CGNs were plated in 10-cm dishes and subjected to OGD treatment for 90 min. Individual plates were reperfused for either 2, 6, or 10 h with regular culture media. At these time points, the cells were scraped from the plate and pelleted, and total protein extracts were collected for RNA or protein extraction to detect changes in NRP-1 expression. (D) For NRP-1 inhibitor studies, CGNs were preincubated for 30 min with or without 0.5 μg/ml of synthetic peptide inhibitor. The cells were subsequently subjected to 90 min of OGD and reperfused for 24 h, at which point the cells were fixed with 4% fresh paraformaldehyde and double immunostained for DAPI and MAP2. Cell death was quantified by manual counting of condensed DAPI-stained nuclei, and the induction in neuronal death (n-fold) was determined.
, P < 0.01 by ANOVA and post hoc Tukey's test. (C) CGNs were plated in 10-cm dishes and subjected to OGD treatment for 90 min. Individual plates were reperfused for either 2, 6, or 10 h with regular culture media. At these time points, the cells were scraped from the plate and pelleted, and total protein extracts were collected for RNA or protein extraction to detect changes in NRP-1 expression. (D) For NRP-1 inhibitor studies, CGNs were preincubated for 30 min with or without 0.5 μg/ml of synthetic peptide inhibitor. The cells were subsequently subjected to 90 min of OGD and reperfused for 24 h, at which point the cells were fixed with 4% fresh paraformaldehyde and double immunostained for DAPI and MAP2. Cell death was quantified by manual counting of condensed DAPI-stained nuclei, and the induction in neuronal death (n-fold) was determined.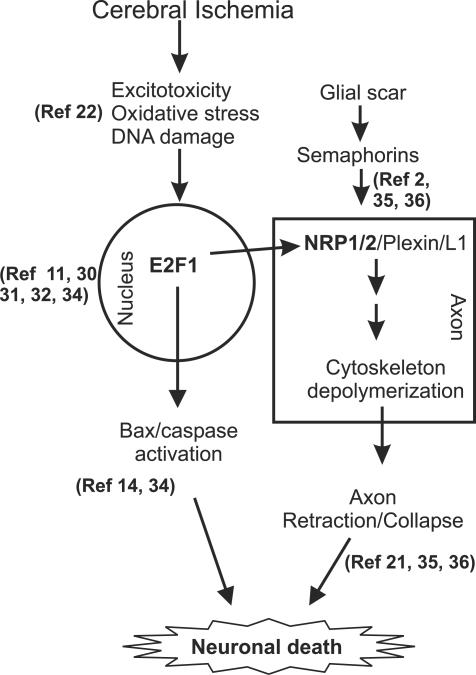
Similar articles
-
The proapoptotic gene SIVA is a direct transcriptional target for the tumor suppressors p53 and E2F1.J Biol Chem. 2004 Jul 2;279(27):28706-14. doi: 10.1074/jbc.M400376200. Epub 2004 Apr 22. J Biol Chem. 2004. PMID: 15105421
-
Transcriptional regulation of the mouse PNRC2 promoter by the nuclear factor Y (NFY) and E2F1.Gene. 2005 Nov 21;361:89-100. doi: 10.1016/j.gene.2005.07.012. Epub 2005 Sep 21. Gene. 2005. PMID: 16181749
-
E2F-HDAC complexes negatively regulate the tumor suppressor gene ARHI in breast cancer.Oncogene. 2006 Jan 12;25(2):230-9. doi: 10.1038/sj.onc.1209025. Oncogene. 2006. PMID: 16158053
-
The regulatory and enzymatic functions of CRMPs in neuritogenesis, synaptic plasticity, and gene transcription.Neurochem Int. 2020 Oct;139:104795. doi: 10.1016/j.neuint.2020.104795. Epub 2020 Jul 8. Neurochem Int. 2020. PMID: 32652266 Review.
-
Recent advances in adenovirus-mediated gene therapy for cerebral ischemia.Curr Gene Ther. 2003 Feb;3(1):43-8. doi: 10.2174/1566523033347516. Curr Gene Ther. 2003. PMID: 12553534 Review.
Cited by
-
Molecular blockade of VEGFR2 in human epithelial ovarian carcinoma cells.Lab Invest. 2010 May;90(5):709-23. doi: 10.1038/labinvest.2010.52. Epub 2010 Mar 1. Lab Invest. 2010. PMID: 20195243 Free PMC article.
-
E2F1 localizes predominantly to neuronal cytoplasm and fails to induce expression of its transcriptional targets in human immunodeficiency virus-induced neuronal damage.Neurosci Lett. 2010 Jul 26;479(2):97-101. doi: 10.1016/j.neulet.2010.05.032. Epub 2010 May 16. Neurosci Lett. 2010. PMID: 20580656 Free PMC article.
-
Loss of non-motor kinesin KIF26A causes congenital brain malformations via dysregulated neuronal migration and axonal growth as well as apoptosis.Dev Cell. 2022 Oct 24;57(20):2381-2396.e13. doi: 10.1016/j.devcel.2022.09.011. Epub 2022 Oct 12. Dev Cell. 2022. PMID: 36228617 Free PMC article.
-
AP-2α-Mediated Activation of E2F and EZH2 Drives Melanoma Metastasis.Cancer Res. 2021 Sep 1;81(17):4455-4470. doi: 10.1158/0008-5472.CAN-21-0772. Epub 2021 Jul 1. Cancer Res. 2021. PMID: 34210752 Free PMC article.
-
Effects of endurance training on the expression of host proteins involved in SARS-CoV-2 cell entry in C57BL/6J mouse.Physiol Rep. 2021 Sep;9(17):e15014. doi: 10.14814/phy2.15014. Physiol Rep. 2021. PMID: 34523264 Free PMC article.
References
-
- Barrett, T., C. Cheadle, W. B. Wood, D. Teichberg, D. M. Donovan, W. J. Freed, K. G. Becker, and M. P. Vawter. 2001. Assembly and use of a broadly applicable neural cDNA microarray. Restor. Neurol. Neurosci. 18:127-135. - PubMed
-
- Beck, H., T. Acker, A. W. Puschel, H. Fujisawa, P. Carmeliet, and K. H. Plate. 2002. Cell type-specific expression of neuropilins in an MCA-occlusion model in mice suggests a potential role in post-ischemic brain remodeling. J. Neuropathol. Exp. Neurol. 61:339-350. - PubMed
-
- Carmichael, S. T. 2006. Cellular and molecular mechanisms of neural repair after stroke: making waves. Ann. Neurol. 59:735-742. - PubMed
-
- Carmichael, S. T., I. Archibeque, L. Luke, T. Nolan, J. Momiy, and S. Li. 2005. Growth-associated gene expression after stroke: evidence for a growth-promoting region in peri-infarct cortex. Exp. Neurol. 193:291-311. - PubMed
-
- Castellani, V. 2002. The function of neuropilin/L1 complex. Adv. Exp. Med. Biol. 515:91-102. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous