

doi: 10.1186/gb-2006-7-11-r104.
Predicting domain-domain interactions using a parsimony approach
Affiliations
- PMID: 17094802
- PMCID: PMC1794579
- DOI: 10.1186/gb-2006-7-11-r104
Item in Clipboard
Predicting domain-domain interactions using a parsimony approach
Genome Biol.
2006.
Abstract
We propose a novel approach to predict domain-domain interactions from a protein-protein interaction network. In our method we apply a parsimony-driven explanation of the network, where the domain interactions are inferred using linear programming optimization, and false positives in the protein network are handled by a probabilistic construction. This method outperforms previous approaches by a considerable margin. The results indicate that the parsimony principle provides a correct approach for detecting domain-domain contacts.
Figures
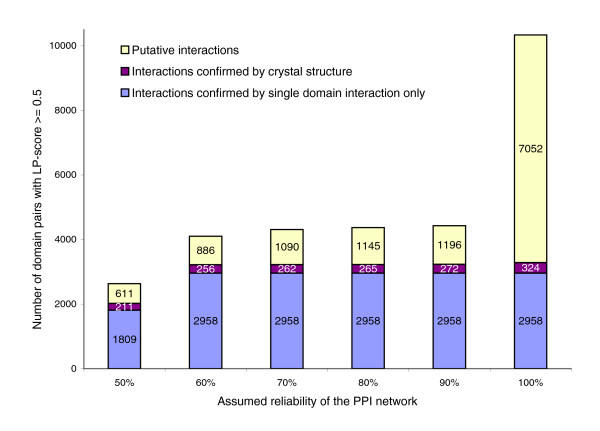
Influence of assumed network reliability on LP-score predictions. Influence of the assumed network reliability on the number of pairs with LP-score above 0.5 and the number of interactions among those that are confirmed by crystal structures in our gold standard set or by witnesses. The number of pairs confirmed by the gold standard set remains stable for all network reliability assumptions, and interactions predicted under assumption of a lower network reliability almost always are a subset of the interactions predicted under the assumption of a higher network reliability.
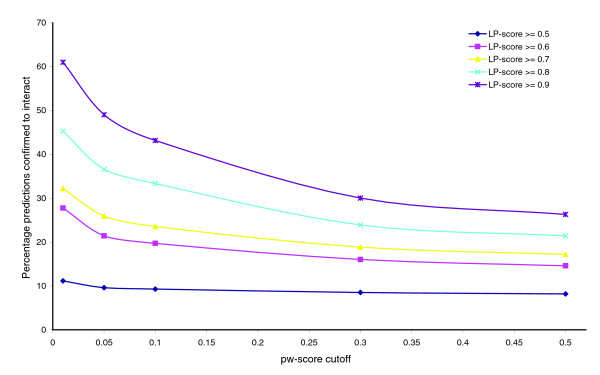
Influence of pw-score cutoff on accuracy of predictions. A pw-score close to 1 indicates a promiscuous domain pair that can obtain a high LP-score independent of the topology of the underlying protein-protein interaction network, and does not have significant witness support. Higher LP-score cutoffs lead to higher prediction accuracy; smaller (more stringent) pw-score cutoffs help improve it further.
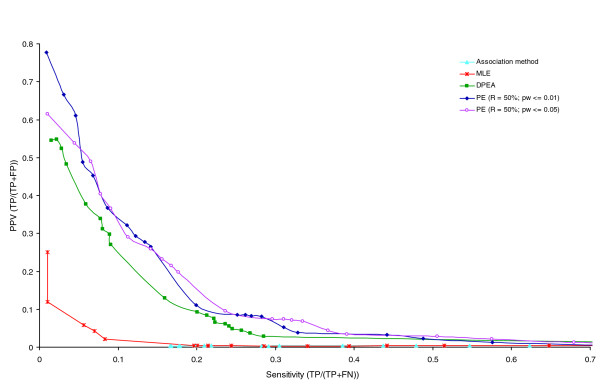
PPV versus sensitivity in enrichment of confirmed interactions experiment. Comparison of PPV (TP/(TP + FP)) and Sensitivity (TP/(TP + FN)) attained by the PE method with pw-score cutoffs of 0.01 and 0.05, and previously by the Association, EM, and DPEA methods. The comparison is based on estimations of how many of the high-scoring domain-domain interactions are confirmed by the gold standard set.
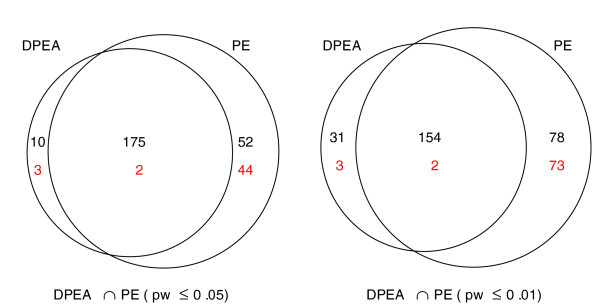
Comparison of gold standard pairs recovered by PE and DPEA. Comparison between the sets of gold standard pairs recovered among the 3,005 pairs considered as high-confidence predictions of the DPEA method and among the 3,000 top scoring pairs selected by the PE method with pw-score cutoffs of 0.01 and 0.05. In red are the numbers of difficult gold standard pairs predicted. In the set of 185 gold standard interactions recovered among the 3,005 high-confidence domain pairs by the DPEA method, only 5 are in the difficult category. In comparison, among the 3,000 top scoring domain interactions reported by the PE method with a pw-score cutoff of 0.05, there are 46 difficult pairs (75 difficult pairs with cutoff 0.01).
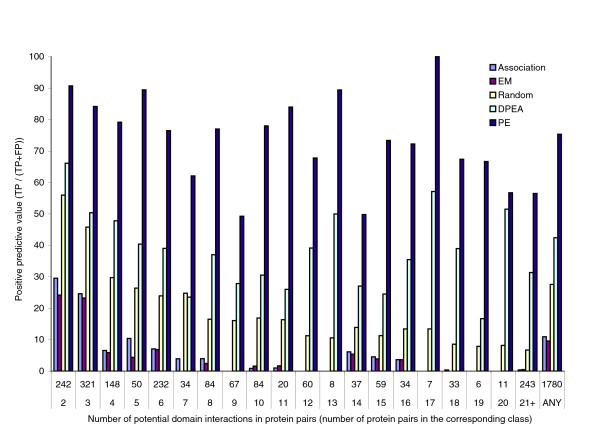
Comparison of positive predictive values in mediating domain pair prediction experiment. Estimated positive predictive value of the Association, EM, PE, and DPEA methods, and the performance expected by chance in such experiments, called the Random method. The results are presented according to the number of potential domain-domain interactions between the protein pairs in the set, and also in general. The numbers along the x-axis represent the number of protein pairs in the corresponding class. The PE method outperforms the previous methods in every class. In particular, for the 242 protein pairs with only 2 potential domain-domain interactions, PE has a PPV of 90.7%, and sensitivity of 93.8%, and for the 993 protein pairs with 2 to 6 potential domain-domain interactions, the PE method consistently has an average PPV above 76%. Overall, the PE method has an estimated average PPV of 75.3%. The Association and the EM methods both perform worse than Random; possible reasons for such an outcome are discussed in the text.
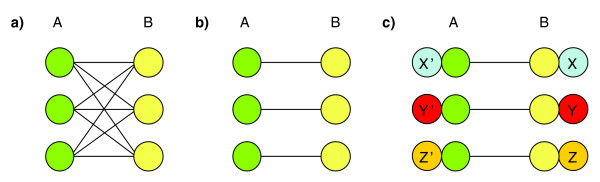
Specificity of interactions. (a) A hypothetical subnetwork for non-specific interaction between proteins containing two domains: each protein containing domain A interacts with each protein containing domain B. Detecting such interactions is easy for all four methods: Association, EM, DPEA, and PE. (b) A hypothetical subnetwork for highly specific interactions between proteins containing domain A and proteins containing domain B. Since only a small number of interactions actually occur, out of all possible interactions between pairs of proteins containing domain pair {A, B}, detecting such specific interactions is difficult for the EM and the Association methods, but not for the DPEA and the PE methods. (c) Hypothetical subnetwork for highly specific interactions in the context of multidomain proteins. PE will attribute these interactions to domain pair {A, B}, as it requires prediction of one interaction {A, B} to justify three protein-protein interactions. On the other hand, the association and the EM method will assign higher probability to domain pairs {X, X'}, {Y, Y'}, and {Z, Z'}, as it is beneficial to assign higher probabilities to interactions involving rare domains, that is, X, Y, and Z.
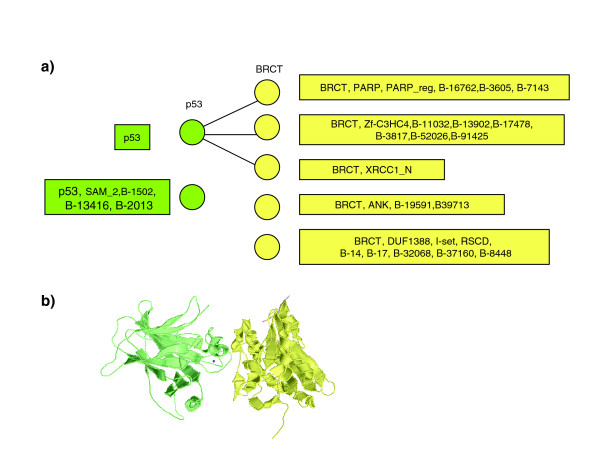
P53-BRCT interactions. (a) The subnetwork of the protein-protein interaction network spanning only the human proteins with p53 and BCRT domains. Three pairs of these proteins interact (as indicated by connecting edges). The domain composition of each protein is given in the corresponding box. PE correctly identifies BRCT-p53 as interacting partners. (b) Crystal structure of the p53-BCRT complex (PDB entry 1gzh); only the p53 and BRCT domains are shown in the figure.
Similar articles
-
Interrogating domain-domain interactions with parsimony based approaches.BMC Bioinformatics. 2008 Mar 26;9:171. doi: 10.1186/1471-2105-9-171. BMC Bioinformatics. 2008. PMID: 18366803 Free PMC article.
-
Inferring protein domain interactions from databases of interacting proteins.Genome Biol. 2005;6(10):R89. doi: 10.1186/gb-2005-6-10-r89. Epub 2005 Sep 19. Genome Biol. 2005. PMID: 16207360 Free PMC article.
-
A discriminative approach for identifying domain-domain interactions from protein-protein interactions.Proteins. 2010 Apr;78(5):1243-53. doi: 10.1002/prot.22643. Proteins. 2010. PMID: 20027642
-
Topology and weights in a protein domain interaction network--a novel way to predict protein interactions.BMC Genomics. 2006 May 23;7:122. doi: 10.1186/1471-2164-7-122. BMC Genomics. 2006. PMID: 16716232 Free PMC article.
-
Kernel methods for predicting protein-protein interactions.Bioinformatics. 2005 Jun;21 Suppl 1:i38-46. doi: 10.1093/bioinformatics/bti1016. Bioinformatics. 2005. PMID: 15961482
Cited by
-
Translating Mendelian and complex inheritance of Alzheimer's disease genes for predicting unique personal genome variants.J Am Med Inform Assoc. 2012 Mar-Apr;19(2):306-16. doi: 10.1136/amiajnl-2011-000656. J Am Med Inform Assoc. 2012. PMID: 22319180 Free PMC article.
-
A top-down approach to infer and compare domain-domain interactions across eight model organisms.PLoS One. 2009;4(3):e5096. doi: 10.1371/journal.pone.0005096. Epub 2009 Mar 31. PLoS One. 2009. PMID: 19333396 Free PMC article.
-
DASMI: exchanging, annotating and assessing molecular interaction data.Bioinformatics. 2009 May 15;25(10):1321-8. doi: 10.1093/bioinformatics/btp142. Bioinformatics. 2009. PMID: 19420069 Free PMC article.
-
Interrogating domain-domain interactions with parsimony based approaches.BMC Bioinformatics. 2008 Mar 26;9:171. doi: 10.1186/1471-2105-9-171. BMC Bioinformatics. 2008. PMID: 18366803 Free PMC article.
-
NetGrep: fast network schema searches in interactomes.Genome Biol. 2008;9(9):R138. doi: 10.1186/gb-2008-9-9-r138. Epub 2008 Sep 18. Genome Biol. 2008. PMID: 18801179 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
