

Mosaic structure of human coronavirus NL63, one thousand years of evolution
- PMID: 17054987
- PMCID: PMC7094706
- DOI: 10.1016/j.jmb.2006.09.074
Mosaic structure of human coronavirus NL63, one thousand years of evolution
Abstract
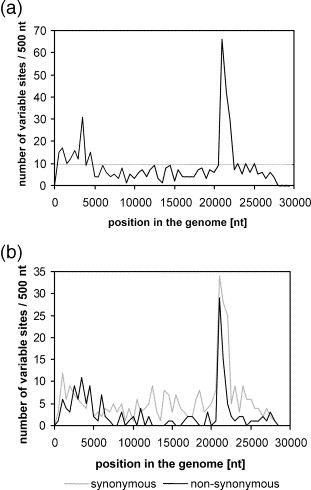
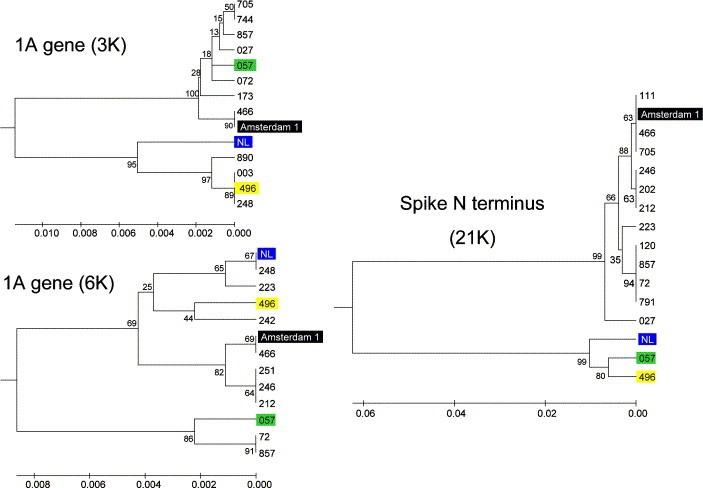
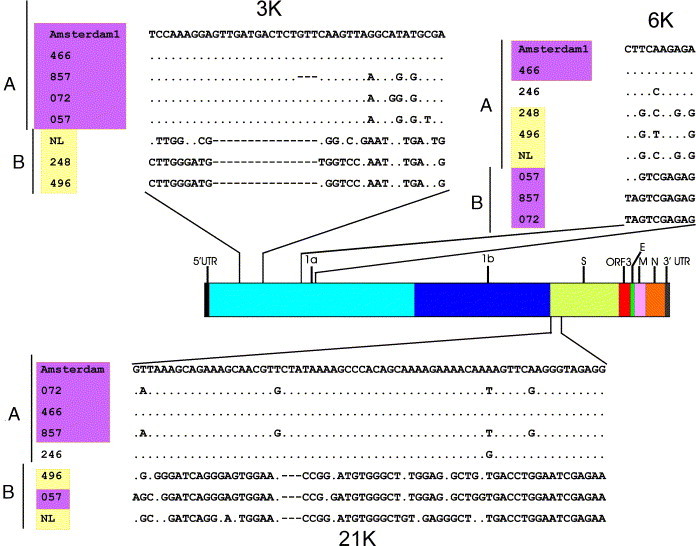
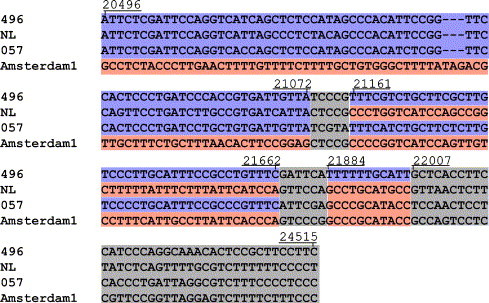
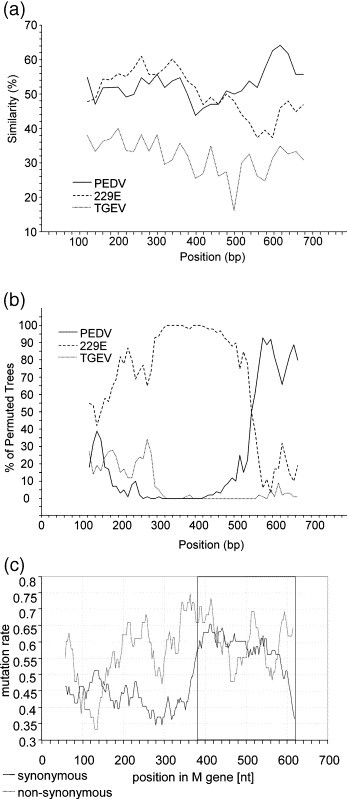
Before the SARS outbreak only two human coronaviruses (HCoV) were known: HCoV-OC43 and HCoV-229E. With the discovery of SARS-CoV in 2003, a third family member was identified. Soon thereafter, we described the fourth human coronavirus (HCoV-NL63), a virus that has spread worldwide and is associated with croup in children. We report here the complete genome sequence of two HCoV-NL63 clinical isolates, designated Amsterdam 57 and Amsterdam 496. The genomes are 27,538 and 27,550 nucleotides long, respectively, and share the same genome organization. We identified two variable regions, one within the 1a and one within the S gene, whereas the 1b and N genes were most conserved. Phylogenetic analysis revealed that HCoV-NL63 genomes have a mosaic structure with multiple recombination sites. Additionally, employing three different algorithms, we assessed the evolutionary rate for the S gene of group Ib coronaviruses to be approximately 3 x 10(-4) substitutions per site per year. Using this evolutionary rate we determined that HCoV-NL63 diverged in the 11th century from its closest relative HCoV-229E.
Figures
Similar articles
-
Epidemiology and clinical presentations of human coronavirus NL63 infections in hong kong children.J Clin Microbiol. 2009 Nov;47(11):3486-92. doi: 10.1128/JCM.00832-09. Epub 2009 Sep 16. J Clin Microbiol. 2009. PMID: 19759228 Free PMC article.
-
Genetic variability of human coronavirus OC43-, 229E-, and NL63-like strains and their association with lower respiratory tract infections of hospitalized infants and immunocompromised patients.J Med Virol. 2006 Jul;78(7):938-49. doi: 10.1002/jmv.20645. J Med Virol. 2006. PMID: 16721849 Free PMC article.
-
Coronavirus HKU1 and other coronavirus infections in Hong Kong.J Clin Microbiol. 2006 Jun;44(6):2063-71. doi: 10.1128/JCM.02614-05. J Clin Microbiol. 2006. PMID: 16757599 Free PMC article.
-
Human coronaviruses: what do they cause?Antivir Ther. 2007;12(4 Pt B):651-8. Antivir Ther. 2007. PMID: 17944272 Review.
-
Human coronavirus NL63, a new respiratory virus.FEMS Microbiol Rev. 2006 Sep;30(5):760-73. doi: 10.1111/j.1574-6976.2006.00032.x. FEMS Microbiol Rev. 2006. PMID: 16911043 Free PMC article. Review.
Cited by
-
Current understanding of middle east respiratory syndrome coronavirus infection in human and animal models.J Thorac Dis. 2018 Jul;10(Suppl 19):S2260-S2271. doi: 10.21037/jtd.2018.03.80. J Thorac Dis. 2018. PMID: 30116605 Free PMC article. Review.
-
Human coronavirus NL63 open reading frame 3 encodes a virion-incorporated N-glycosylated membrane protein.Virol J. 2010 Jan 15;7:6. doi: 10.1186/1743-422X-7-6. Virol J. 2010. PMID: 20078868 Free PMC article.
-
Evaluating the Virology and Evolution of Seasonal Human Coronaviruses Associated with the Common Cold in the COVID-19 Era.Microorganisms. 2023 Feb 10;11(2):445. doi: 10.3390/microorganisms11020445. Microorganisms. 2023. PMID: 36838410 Free PMC article. Review.
-
Origins and Evolution of Seasonal Human Coronaviruses.Viruses. 2022 Jul 15;14(7):1551. doi: 10.3390/v14071551. Viruses. 2022. PMID: 35891531 Free PMC article.
-
Antibody escape, the risk of serotype formation, and rapid immune waning: Modeling the implications of SARS-CoV-2 immune evasion.PLoS One. 2023 Oct 18;18(10):e0292099. doi: 10.1371/journal.pone.0292099. eCollection 2023. PLoS One. 2023. PMID: 37851632 Free PMC article.
References
-
- Lai M.M.C., Holmes K.V. Coronaviridae and their replication. In: Howley P., Griffin D., Lamb R., Martin M., Roizman B., Straus S., Knipe D., editors. Fields-Virology. Lippincott Williams and Wilkins; London: 2005. pp. 1163–1185.
-
- Cavanagh D., Brian D.A., Brinton M.A., Enjuanes L., Holmes K.V., Horzinek M.C. The Coronaviridae now comprises two genera, coronavirus and torovirus: report of the Coronaviridae Study Group. Adv. Exp. Med. Biol. 1993;342:255–257. - PubMed
Publication types
MeSH terms
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
