

Sequence-specific solvent accessibilities of protein residues in unfolded protein ensembles
- PMID: 17012314
- PMCID: PMC1779920
- DOI: 10.1529/biophysj.106.087528
Sequence-specific solvent accessibilities of protein residues in unfolded protein ensembles
Abstract
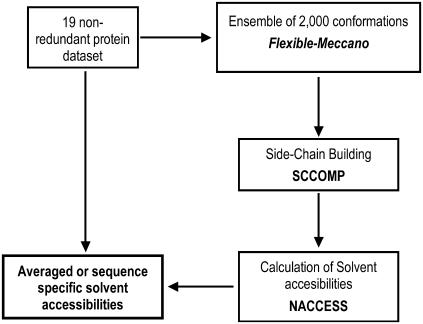
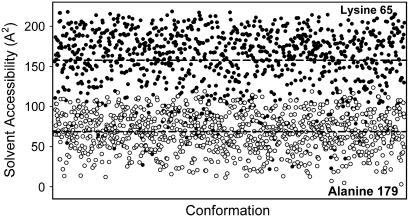
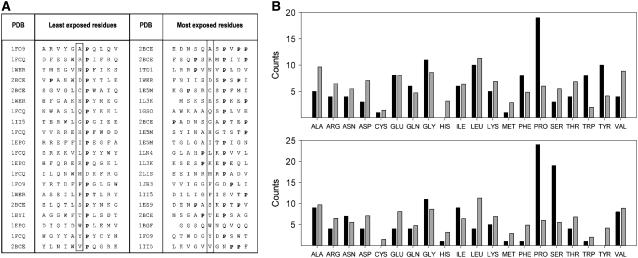
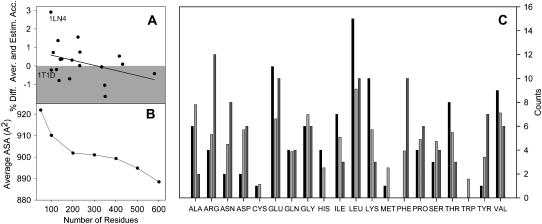
Protein stability cannot be understood without the correct description of the unfolded state. We present here an efficient method for accurate calculation of atomic solvent exposures for denatured protein ensembles. The method used to generate the ensembles has been shown to reproduce diverse biophysical experimental data corresponding to natively and chemically unfolded proteins. Using a data set of 19 nonhomologous proteins containing from 98 to 579 residues, we report average accessibilities for all residue types. These averaged accessibilities are considerably lower than those previously reported for tripeptides and close to the lower limit reported by Creamer and co-workers. Of importance, we observe remarkable sequence dependence for the exposure to solvent of all residue types, which indicates that average residue solvent exposures can be inappropriate to interpret mutational studies. In addition, we observe smaller influences of both protein size and protein amino acid composition in the averaged residue solvent exposures for individual proteins. Calculating residue-specific solvent accessibilities within the context of real sequences is thus necessary and feasible. The approach presented here may allow a more precise parameterization of protein energetics as a function of polar- and apolar-area burial and opens new ways to investigate the energetics of the unfolded state of proteins.
Figures
Similar articles
-
ProtSA: a web application for calculating sequence specific protein solvent accessibilities in the unfolded ensemble.BMC Bioinformatics. 2009 Apr 8;10:104. doi: 10.1186/1471-2105-10-104. BMC Bioinformatics. 2009. PMID: 19356231 Free PMC article.
-
Alpha-helix stabilization by alanine relative to glycine: roles of polar and apolar solvent exposures and of backbone entropy.Proteins. 2006 Aug 15;64(3):769-78. doi: 10.1002/prot.21041. Proteins. 2006. PMID: 16755589
-
Residue solvent accessibilities in the unfolded polypeptide chain.Biophys J. 1992 Dec;63(6):1483-6. doi: 10.1016/S0006-3495(92)81746-0. Biophys J. 1992. PMID: 1489908 Free PMC article.
-
Protein structure, stability and solubility in water and other solvents.Philos Trans R Soc Lond B Biol Sci. 2004 Aug 29;359(1448):1225-34; discussion 1234-5. doi: 10.1098/rstb.2004.1500. Philos Trans R Soc Lond B Biol Sci. 2004. PMID: 15306378 Free PMC article. Review.
-
Atom depth in protein structure and function.Trends Biochem Sci. 2003 Nov;28(11):593-7. doi: 10.1016/j.tibs.2003.09.004. Trends Biochem Sci. 2003. PMID: 14607089 Review.
Cited by
-
A self-consistent description of the conformational behavior of chemically denatured proteins from NMR and small angle scattering.Biophys J. 2009 Nov 18;97(10):2839-45. doi: 10.1016/j.bpj.2009.08.044. Biophys J. 2009. PMID: 19917239 Free PMC article.
-
Worm-like Ising model for protein mechanical unfolding under the effect of osmolytes.Biophys J. 2012 Jan 18;102(2):342-50. doi: 10.1016/j.bpj.2011.12.007. Biophys J. 2012. PMID: 22339871 Free PMC article.
-
Why Hofmeister effects of many salts favor protein folding but not DNA helix formation.Proc Natl Acad Sci U S A. 2010 Apr 27;107(17):7716-21. doi: 10.1073/pnas.0913376107. Epub 2010 Apr 12. Proc Natl Acad Sci U S A. 2010. PMID: 20385834 Free PMC article.
-
On methods for determining solvent accessible surface area for proteins in their unfolded state.BMC Res Notes. 2014 Sep 3;7:602. doi: 10.1186/1756-0500-7-602. BMC Res Notes. 2014. PMID: 25187400 Free PMC article.
-
In silico Evaluation of Crosslinking Effects on Denaturant m(eq) values and ΔCp upon Protein Unfolding.Avicenna J Med Biotechnol. 2012 Jan;4(1):23-34. Avicenna J Med Biotechnol. 2012. PMID: 23408172 Free PMC article.
References
-
- Shortle, D. 1996. The denatured state (the other half of the folding equation) and its role in protein stability. FASEB J. 10:27–34. - PubMed
-
- Funahashi, J., Y. Sugita, A. Kitao, and K. Yutani. 2003. How can free energy component analysis explain the difference in protein stability caused by amino acid substitutions? Effect of three hydrophobic mutations at the 56th residue on the stability of human lysozyme. Protein Eng. Des. Sel. 16:665–671. - PubMed
-
- Lazaridis, T., and M. Karplus. 2003. Thermodynamics of protein folding: a microscopic view. Biophys. Chem. 100:367–395. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
