

Expression-guided in silico evaluation of candidate cis regulatory codes for Drosophila muscle founder cells
- PMID: 16733548
- PMCID: PMC1464814
- DOI: 10.1371/journal.pcbi.0020053
Expression-guided in silico evaluation of candidate cis regulatory codes for Drosophila muscle founder cells
Abstract
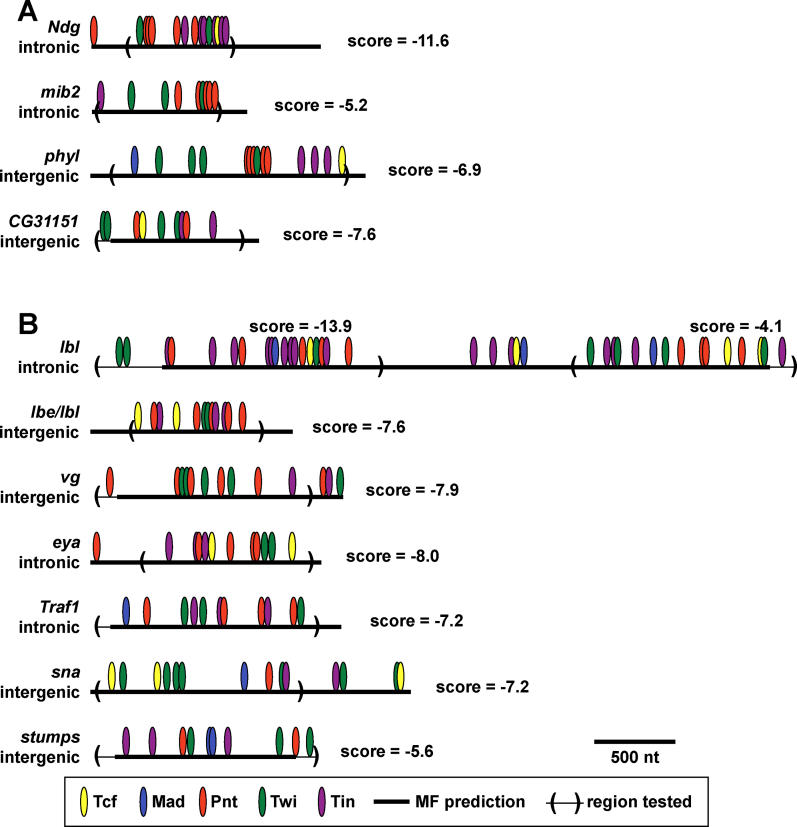
While combinatorial models of transcriptional regulation can be inferred for metazoan systems from a priori biological knowledge, validation requires extensive and time-consuming experimental work. Thus, there is a need for computational methods that can evaluate hypothesized cis regulatory codes before the difficult task of experimental verification is undertaken. We have developed a novel computational framework (termed "CodeFinder") that integrates transcription factor binding site and gene expression information to evaluate whether a hypothesized transcriptional regulatory model (TRM; i.e., a set of co-regulating transcription factors) is likely to target a given set of co-expressed genes. Our basic approach is to simultaneously predict cis regulatory modules (CRMs) associated with a given gene set and quantify the enrichment for combinatorial subsets of transcription factor binding site motifs comprising the hypothesized TRM within these predicted CRMs. As a model system, we have examined a TRM experimentally demonstrated to drive the expression of two genes in a sub-population of cells in the developing Drosophila mesoderm, the somatic muscle founder cells. This TRM was previously hypothesized to be a general mode of regulation for genes expressed in this cell population. In contrast, the present analyses suggest that a modified form of this cis regulatory code applies to only a subset of founder cell genes, those whose gene expression responds to specific genetic perturbations in a similar manner to the gene on which the original model was based. We have confirmed this hypothesis by experimentally discovering six (out of 12 tested) new CRMs driving expression in the embryonic mesoderm, four of which drive expression in founder cells.
Conflict of interest statement
Figures








Similar articles
-
Developmental regulation of the Drosophila Tropomyosin I (TmI) gene is controlled by a muscle activator enhancer region that contains multiple cis-elements and binding sites for multiple proteins.Dev Genet. 1997;20(4):297-306. doi: 10.1002/(SICI)1520-6408(1997)20:4<297::AID-DVG1>3.0.CO;2-2. Dev Genet. 1997. PMID: 9254904
-
De novo prediction of cis-regulatory elements and modules through integrative analysis of a large number of ChIP datasets.BMC Genomics. 2014 Dec 2;15:1047. doi: 10.1186/1471-2164-15-1047. BMC Genomics. 2014. PMID: 25442502 Free PMC article.
-
Identification of cis-regulatory modules encoding temporal dynamics during development.BMC Genomics. 2014 Jun 27;15(1):534. doi: 10.1186/1471-2164-15-534. BMC Genomics. 2014. PMID: 24972496 Free PMC article.
-
Developmental mechanisms and cis-regulatory codes.Curr Opin Genet Dev. 2006 Apr;16(2):165-70. doi: 10.1016/j.gde.2006.02.014. Epub 2006 Feb 28. Curr Opin Genet Dev. 2006. PMID: 16503128 Review.
-
Diversification of muscle types in Drosophila: upstream and downstream of identity genes.Curr Top Dev Biol. 2012;98:277-301. doi: 10.1016/B978-0-12-386499-4.00011-2. Curr Top Dev Biol. 2012. PMID: 22305167 Review.
Cited by
-
Proteogenomic analysis of psoriasis reveals discordant and concordant changes in mRNA and protein abundance.Genome Med. 2015 Aug 4;7(1):86. doi: 10.1186/s13073-015-0208-5. eCollection 2015. Genome Med. 2015. PMID: 26251673 Free PMC article.
-
Org-1, the Drosophila ortholog of Tbx1, is a direct activator of known identity genes during muscle specification.Development. 2012 Mar;139(5):1001-12. doi: 10.1242/dev.073890. Development. 2012. PMID: 22318630 Free PMC article.
-
The complex spatio-temporal regulation of the Drosophila myoblast attractant gene duf/kirre.PLoS One. 2009 Sep 9;4(9):e6960. doi: 10.1371/journal.pone.0006960. PLoS One. 2009. PMID: 19742310 Free PMC article.
-
ModuleMiner - improved computational detection of cis-regulatory modules: are there different modes of gene regulation in embryonic development and adult tissues?Genome Biol. 2008 Apr 7;9(4):R66. doi: 10.1186/gb-2008-9-4-r66. Genome Biol. 2008. PMID: 18394174 Free PMC article.
-
Identification of receptor-tyrosine-kinase-signaling target genes reveals receptor-specific activities and pathway branchpoints during Drosophila development.Genetics. 2009 Apr;181(4):1335-45. doi: 10.1534/genetics.108.098475. Epub 2009 Feb 2. Genetics. 2009. PMID: 19189950 Free PMC article.
References
-
- Davidson EH. Genomic regulatory systems. San Diego (California): Academic Press; 2001. 261. p.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
