

Postnatal acquisition of endotoxin tolerance in intestinal epithelial cells
- PMID: 16606665
- PMCID: PMC2118301
- DOI: 10.1084/jem.20050625
Postnatal acquisition of endotoxin tolerance in intestinal epithelial cells
Abstract
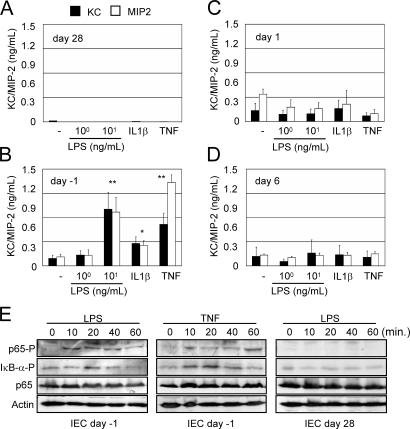
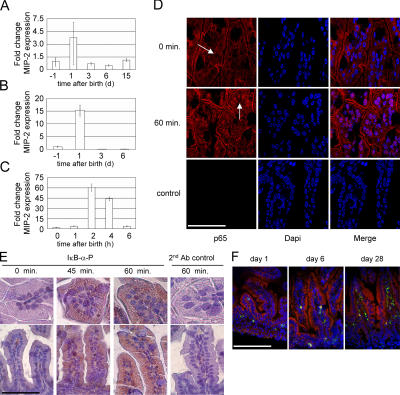
The role of innate immune recognition by intestinal epithelial cells (IECs) in vivo is ill-defined. Here, we used highly enriched primary IECs to analyze Toll-like receptor (TLR) signaling and mechanisms that prevent inappropriate stimulation by the colonizing microflora. Although the lipopolysaccharide (LPS) receptor complex TLR4/MD-2 was present in fetal, neonatal, and adult IECs, LPS-induced nuclear factor kappaB (NF-kappaB) activation and chemokine (macrophage inflammatory protein 2 [MIP-2]) secretion was only detected in fetal IECs. Fetal intestinal macrophages, in contrast, were constitutively nonresponsive to LPS. Acquisition of LPS resistance was paralleled by a spontaneous activation of IECs shortly after birth as illustrated by phosphorylation of IkappaB-alpha and nuclear translocation of NF-kappaB p65 in situ as well as transcriptional activation of MIP-2. Importantly, the spontaneous IEC activation occurred in vaginally born mice but not in neonates delivered by Caesarean section or in TLR4-deficient mice, which together with local endotoxin measurements identified LPS as stimulatory agent. The postnatal loss of LPS responsiveness of IECs was associated with a posttranscriptional down-regulation of the interleukin 1 receptor-associated kinase 1, which was essential for epithelial TLR4 signaling in vitro. Thus, unlike intestinal macrophages, IECs acquire TLR tolerance immediately after birth by exposure to exogenous endotoxin to facilitate microbial colonization and the development of a stable intestinal host-microbe homeostasis.
Figures







Similar articles
-
Regulation of Toll-like receptor 4-associated MD-2 in intestinal epithelial cells: a comprehensive analysis.Innate Immun. 2010 Apr;16(2):93-103. doi: 10.1177/1753425909339231. Epub 2009 Aug 26. Innate Immun. 2010. PMID: 19710105 Free PMC article.
-
Induction of in vitro reprogramming by Toll-like receptor (TLR)2 and TLR4 agonists in murine macrophages: effects of TLR "homotolerance" versus "heterotolerance" on NF-kappa B signaling pathway components.J Immunol. 2003 Jan 1;170(1):508-19. doi: 10.4049/jimmunol.170.1.508. J Immunol. 2003. PMID: 12496438
-
Funiculosin variants and phosphorylated derivatives promote innate immune responses via the Toll-like receptor 4/myeloid differentiation factor-2 complex.J Biol Chem. 2017 Sep 15;292(37):15378-15394. doi: 10.1074/jbc.M117.791780. Epub 2017 Jul 28. J Biol Chem. 2017. PMID: 28754693 Free PMC article.
-
The "polarizing-tolerizing" mechanism of intestinal epithelium: its relevance to colonic homeostasis.Semin Immunopathol. 2008 Feb;30(1):3-9. doi: 10.1007/s00281-007-0099-7. Epub 2007 Nov 20. Semin Immunopathol. 2008. PMID: 18026955 Review.
-
The Regulation of Endotoxin Tolerance and its Impact on Macrophage Activation.Crit Rev Immunol. 2015;35(4):293-323. doi: 10.1615/critrevimmunol.2015015495. Crit Rev Immunol. 2015. PMID: 26757393 Review.
Cited by
-
Development of systemic and mucosal immune responses against gut microbiota in early life and implications for the onset of allergies.Front Allergy. 2024 Jul 17;5:1439303. doi: 10.3389/falgy.2024.1439303. eCollection 2024. Front Allergy. 2024. PMID: 39086886 Free PMC article. Review.
-
Can Postbiotics Represent a New Strategy for NEC?Adv Exp Med Biol. 2024;1449:43-57. doi: 10.1007/978-3-031-58572-2_3. Adv Exp Med Biol. 2024. PMID: 39060730 Review.
-
Epithelial-derived interleukin-23 promotes oral mucosal immunopathology.Immunity. 2024 Apr 9;57(4):859-875.e11. doi: 10.1016/j.immuni.2024.02.020. Epub 2024 Mar 20. Immunity. 2024. PMID: 38513665
-
The maternal gut microbiome in pregnancy: implications for the developing immune system.Nat Rev Gastroenterol Hepatol. 2024 Jan;21(1):35-45. doi: 10.1038/s41575-023-00864-2. Epub 2023 Dec 14. Nat Rev Gastroenterol Hepatol. 2024. PMID: 38097774 Review.
-
Commensal bacteria weaken the intestinal barrier by suppressing epithelial neuropilin-1 and Hedgehog signaling.Nat Metab. 2023 Jul;5(7):1174-1187. doi: 10.1038/s42255-023-00828-5. Epub 2023 Jul 6. Nat Metab. 2023. PMID: 37414930 Free PMC article.
References
-
- Medzhitov, R. 2001. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 1:135–145. - PubMed
-
- Abreu, M.T., P. Vora, E. Faure, L.S. Thomas, E.T. Arnold, and M. Arditi. 2001. Decreased expression of Toll-like receptor-4 and MD-2 correlates with intestinal epithelial cell protection against dysregulated proinflammatory gene expression in response to bacterial lipopolysaccharide. J. Immunol. 167:1609–1616. - PubMed
-
- Naik, S., E.J. Kelly, L. Meijer, S. Pettersson, and I.R. Sanderson. 2001. Absence of Toll-like receptor 4 explains endotoxin hyporesponsiveness in human intestinal epithelium. J. Pediatr. Gastroenterol. Nutr. 32:449–453. - PubMed
-
- Cario, E., I.M. Rosenberg, S.L. Brandwein, P.L. Beck, H.C. Reinecker, and D.K. Podolsky. 2000. Lipopolysaccharide activates distinct signaling pathways in intestinal epithelial cell lines expressing Toll-like receptors. J. Immunol. 164:966–972. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
