

How corticosteroids control inflammation: Quintiles Prize Lecture 2005
- PMID: 16604091
- PMCID: PMC1751559
- DOI: 10.1038/sj.bjp.0706736
How corticosteroids control inflammation: Quintiles Prize Lecture 2005
Abstract
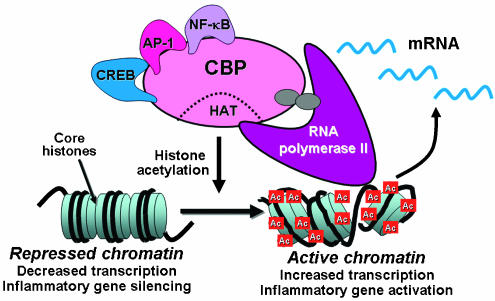
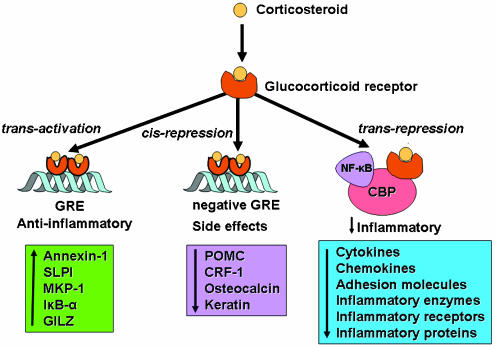
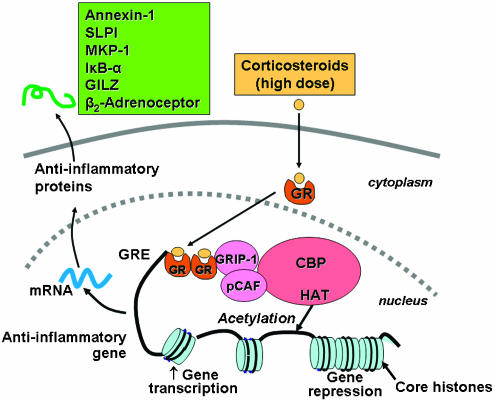
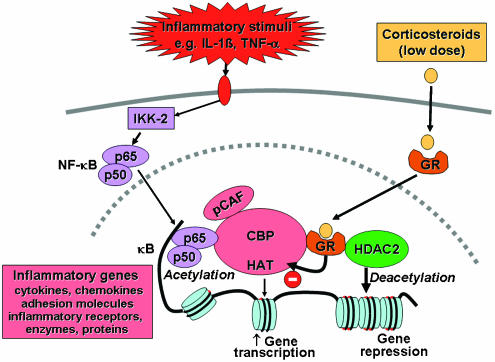
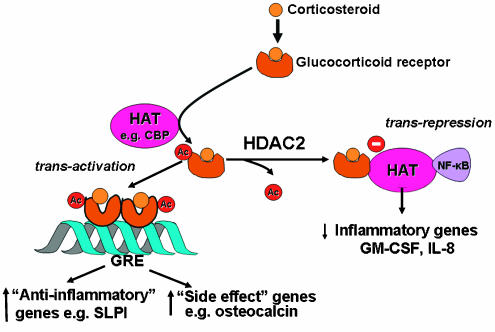
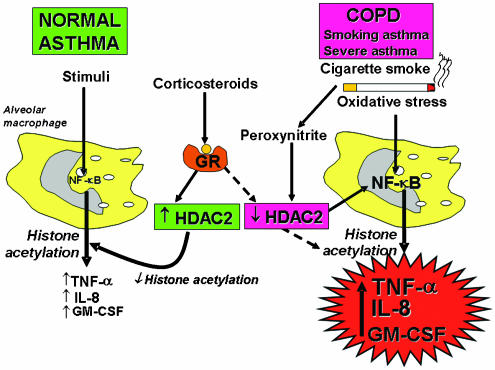
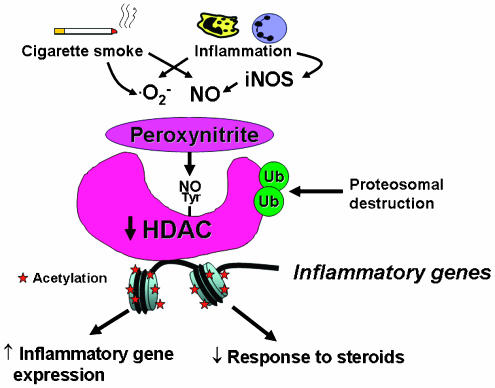
Corticosteroids are the most effective anti-inflammatory therapy for many chronic inflammatory diseases, such as asthma but are relatively ineffective in other diseases such as chronic obstructive pulmonary disease (COPD). Chronic inflammation is characterised by the increased expression of multiple inflammatory genes that are regulated by proinflammatory transcription factors, such as nuclear factor-kappaB and activator protein-1, that bind to and activate coactivator molecules, which then acetylate core histones to switch on gene transcription. Corticosteroids suppress the multiple inflammatory genes that are activated in chronic inflammatory diseases, such as asthma, mainly by reversing histone acetylation of activated inflammatory genes through binding of liganded glucocorticoid receptors (GR) to coactivators and recruitment of histone deacetylase-2 (HDAC2) to the activated transcription complex. At higher concentrations of corticosteroids GR homodimers also interact with DNA recognition sites to active transcription of anti-inflammatory genes and to inhibit transcription of several genes linked to corticosteroid side effects. In patients with COPD and severe asthma and in asthmatic patients who smoke HDAC2 is markedly reduced in activity and expression as a result of oxidative/nitrative stress so that inflammation becomes resistant to the anti-inflammatory actions of corticosteroids. Theophylline, by activating HDAC, may reverse this corticosteroid resistance. This research may lead to the development of novel anti-inflammatory approaches to manage severe inflammatory diseases.
Figures
Similar articles
-
Corticosteroids: the drugs to beat.Eur J Pharmacol. 2006 Mar 8;533(1-3):2-14. doi: 10.1016/j.ejphar.2005.12.052. Epub 2006 Jan 24. Eur J Pharmacol. 2006. PMID: 16436275 Review.
-
Corticosteroid effects on cell signalling.Eur Respir J. 2006 Feb;27(2):413-26. doi: 10.1183/09031936.06.00125404. Eur Respir J. 2006. PMID: 16452600 Review.
-
Histone acetylation and deacetylation: importance in inflammatory lung diseases.Eur Respir J. 2005 Mar;25(3):552-63. doi: 10.1183/09031936.05.00117504. Eur Respir J. 2005. PMID: 15738302 Review.
-
Mechanisms and resistance in glucocorticoid control of inflammation.J Steroid Biochem Mol Biol. 2010 May 31;120(2-3):76-85. doi: 10.1016/j.jsbmb.2010.02.018. Epub 2010 Feb 25. J Steroid Biochem Mol Biol. 2010. PMID: 20188830 Review.
-
Histone deacetylation: an important mechanism in inflammatory lung diseases.COPD. 2005 Dec;2(4):445-55. doi: 10.1080/15412550500346683. COPD. 2005. PMID: 17147010 Review.
Cited by
-
Lactoferrin Supplementation in Preventing and Protecting from SARS-CoV-2 Infection: Is There Any Role in General and Special Populations? An Updated Review of Literature.Int J Mol Sci. 2024 Sep 24;25(19):10248. doi: 10.3390/ijms251910248. Int J Mol Sci. 2024. PMID: 39408576 Free PMC article. Review.
-
Natural products for the treatment of allergic rhinitis: focus on cellular signaling pathways and pharmacological targets.Front Pharmacol. 2024 Sep 30;15:1447097. doi: 10.3389/fphar.2024.1447097. eCollection 2024. Front Pharmacol. 2024. PMID: 39403140 Free PMC article. Review.
-
Molded Round Window Niche Implant as a Dexamethasone Delivery System in a Cochlear Implant-Trauma Animal Model.Pharmaceutics. 2024 Sep 23;16(9):1236. doi: 10.3390/pharmaceutics16091236. Pharmaceutics. 2024. PMID: 39339272 Free PMC article.
-
Molecular, Pathophysiological, and Clinical Aspects of Corticosteroid-Induced Neuropsychiatric Effects: From Bench to Bedside.Biomedicines. 2024 Sep 19;12(9):2131. doi: 10.3390/biomedicines12092131. Biomedicines. 2024. PMID: 39335644 Free PMC article. Review.
-
Physiological Effects of Psychological Interventions Among Persons with Financial Stress: A Systematic Review, Meta-analysis, and Introduction to Psychophysiological Economics.Appl Psychophysiol Biofeedback. 2024 Sep 27. doi: 10.1007/s10484-024-09658-x. Online ahead of print. Appl Psychophysiol Biofeedback. 2024. PMID: 39331272 Review.
References
-
- ADCOCK I.M., COSIO B., TSAPROUNI L., BARNES P.J., ITO K. Redox regulation of histone deacetylases and glucocorticoid-mediated inhibition of the inflammatory response. Antioxid. Redox. Signal. 2005;7:144–152. - PubMed
-
- ANDERSON P., PHILLIPS K., STOECKLIN G., KEDERSHA N. Post-transcriptional regulation of proinflammatory proteins. J. Leukoc. Biol. 2004;76:42–47. - PubMed
-
- BANNISTER A.J., SCHNEIDER R., KOUZARIDES T. Histone methylation: dynamic or static. Cell. 2002;109:801–806. - PubMed
-
- BARNES P.J. Theophylline: new perspectives on an old drug. Am. J. Respir. Crit. Care Med. 2003;167:813–818. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
