

Dystroglycan is selectively cleaved at the parenchymal basement membrane at sites of leukocyte extravasation in experimental autoimmune encephalomyelitis
- PMID: 16585265
- PMCID: PMC2118280
- DOI: 10.1084/jem.20051342
Dystroglycan is selectively cleaved at the parenchymal basement membrane at sites of leukocyte extravasation in experimental autoimmune encephalomyelitis
Abstract
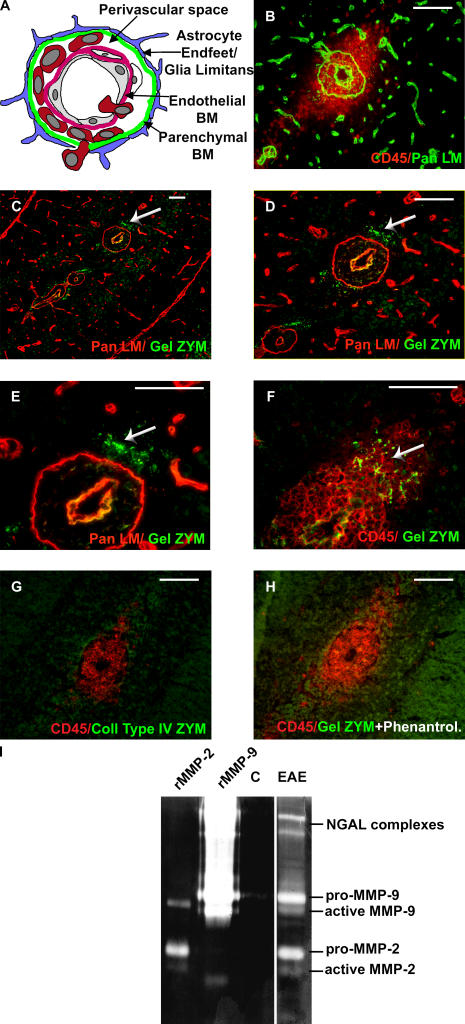
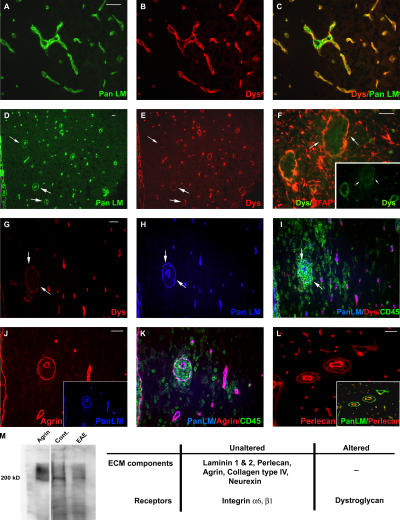
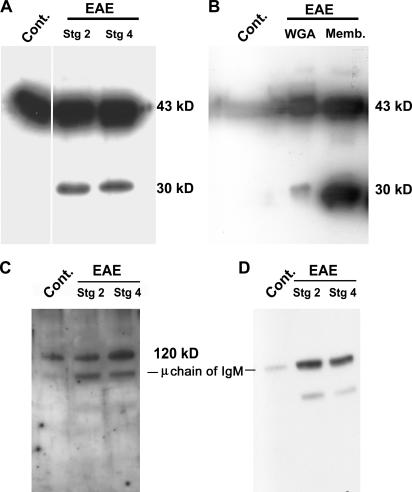
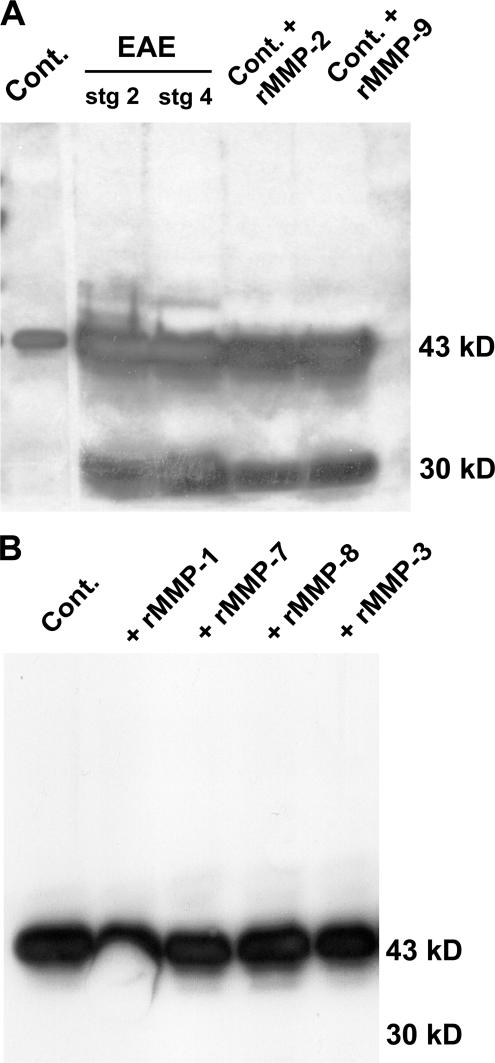
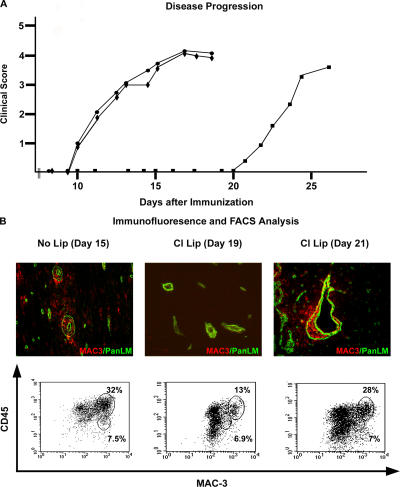
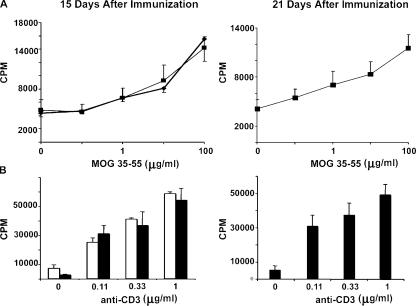
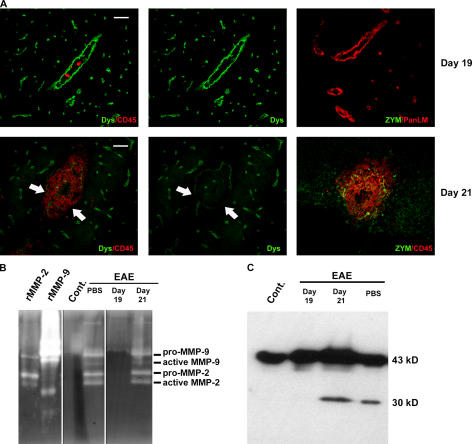
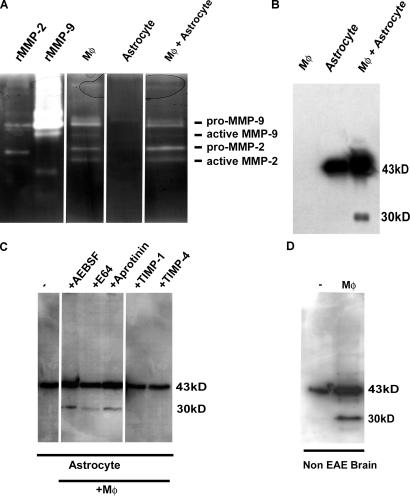
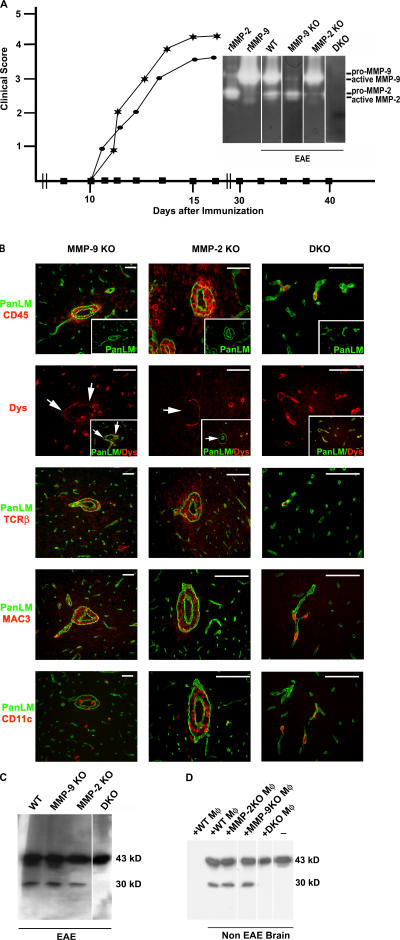
The endothelial cell monolayer of cerebral vessels and its basement membrane (BM) are ensheathed by the astrocyte endfeet, the leptomeningeal cells, and their associated parenchymal BM, all of which contribute to establishment of the blood-brain barrier (BBB). As a consequence of this unique structure, leukocyte penetration of cerebral vessels is a multistep event. In mouse experimental autoimmune encephalomyelitis (EAE), a widely used central nervous system inflammatory model, leukocytes first penetrate the endothelial cell monolayer and underlying BM using integrin beta1-mediated processes, but mechanisms used to penetrate the second barrier defined by the parenchymal BM and glia limitans remain uninvestigated. We show here that macrophage-derived gelatinase (matrix metalloproteinase [MMP]-2 and MMP-9) activity is crucial for leukocyte penetration of the parenchymal BM. Dystroglycan, a transmembrane receptor that anchors astrocyte endfeet to the parenchymal BM via high affinity interactions with laminins 1 and 2, perlecan and agrin, is identified as a specific substrate of MMP-2 and MMP-9. Ablation of both MMP-2 and MMP-9 in double knockout mice confers resistance to EAE by inhibiting dystroglycan cleavage and preventing leukocyte infiltration. This is the first description of selective in situ proteolytic damage of a BBB-specific molecule at sites of leukocyte infiltration.
Figures
Similar articles
-
Focal MMP-2 and MMP-9 activity at the blood-brain barrier promotes chemokine-induced leukocyte migration.Cell Rep. 2015 Feb 24;10(7):1040-54. doi: 10.1016/j.celrep.2015.01.037. Epub 2015 Feb 19. Cell Rep. 2015. PMID: 25704809
-
Loss of astrocyte polarity marks blood-brain barrier impairment during experimental autoimmune encephalomyelitis.Acta Neuropathol. 2009 Aug;118(2):219-33. doi: 10.1007/s00401-009-0558-4. Epub 2009 Jun 17. Acta Neuropathol. 2009. PMID: 19533155
-
Endothelial cell laminin isoforms, laminins 8 and 10, play decisive roles in T cell recruitment across the blood-brain barrier in experimental autoimmune encephalomyelitis.J Cell Biol. 2001 May 28;153(5):933-46. doi: 10.1083/jcb.153.5.933. J Cell Biol. 2001. PMID: 11381080 Free PMC article.
-
The gelatinases, MMP-2 and MMP-9, as fine tuners of neuroinflammatory processes.Matrix Biol. 2019 Jan;75-76:102-113. doi: 10.1016/j.matbio.2017.11.007. Epub 2017 Nov 21. Matrix Biol. 2019. PMID: 29158162 Review.
-
On the structure and functions of gelatinase B/matrix metalloproteinase-9 in neuroinflammation.Prog Brain Res. 2014;214:193-206. doi: 10.1016/B978-0-444-63486-3.00009-8. Prog Brain Res. 2014. PMID: 25410359 Review.
Cited by
-
In vivo methods for imaging blood-brain barrier function and dysfunction.Eur J Nucl Med Mol Imaging. 2023 Mar;50(4):1051-1083. doi: 10.1007/s00259-022-05997-1. Epub 2022 Nov 28. Eur J Nucl Med Mol Imaging. 2023. PMID: 36437425 Free PMC article. Review.
-
The contribution of neutrophils to CNS autoimmunity.Clin Immunol. 2018 Apr;189:23-28. doi: 10.1016/j.clim.2016.06.017. Epub 2016 Jul 1. Clin Immunol. 2018. PMID: 27377536 Free PMC article. Review.
-
New biomarkers for stage determination in Trypanosoma brucei rhodesiense sleeping sickness patients.Clin Transl Med. 2013 Jan 7;2(1):1. doi: 10.1186/2001-1326-2-1. Clin Transl Med. 2013. PMID: 23369533 Free PMC article.
-
Astrocytes in Multiple Sclerosis-Essential Constituents with Diverse Multifaceted Functions.Int J Mol Sci. 2021 May 31;22(11):5904. doi: 10.3390/ijms22115904. Int J Mol Sci. 2021. PMID: 34072790 Free PMC article. Review.
-
The retinal pigment epithelium as a gateway for monocyte trafficking into the eye.EMBO J. 2016 Jun 1;35(11):1219-35. doi: 10.15252/embj.201694202. Epub 2016 Apr 22. EMBO J. 2016. PMID: 27107049 Free PMC article.
References
-
- Wolf, K., R. Muller, S. Borgmann, E.B. Brocker, and P. Friedl. 2003. Amoeboid shape change and contact guidance: T-lymphocyte crawling through fibrillar collagen is independent of matrix remodeling by MMPs and other proteases. Blood. 102:3262–3269. - PubMed
-
- Yadav, R., K.Y. Larbi, R.E. Young, and S. Nourshargh. 2003. Migration of leukocytes through the vessel wall and beyond. Thromb. Haemost. 90:598–606. - PubMed
-
- Engelhardt, B. 1997. Lymphocyte trafficking through the central nervous system. In Adhesion Molecules and Chemokines in Lymphocyte Trafficking. A. Hamann, editor. Harwood Academic Publishers, Amsterdam. 173–200.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
