

Effects of HIV protease inhibitor ritonavir on Akt-regulated cell proliferation in breast cancer
- PMID: 16551874
- PMCID: PMC2727652
- DOI: 10.1158/1078-0432.CCR-05-1167
Effects of HIV protease inhibitor ritonavir on Akt-regulated cell proliferation in breast cancer
Abstract
Purpose: These studies were designed to determine whether ritonavir inhibits breast cancer in vitro and in vivo and, if so, how.
Experimental design: Ritonavir effects on breast cancer cell growth were studied in the estrogen receptor (ER)-positive lines MCF7 and T47D and in the ER-negative lines MDA-MB-436 and MDA-MB-231. Effects of ritonavir on Rb-regulated and Akt-mediated cell proliferation were studied. Ritonavir was tested for inhibition of a mammary carcinoma xenograft.
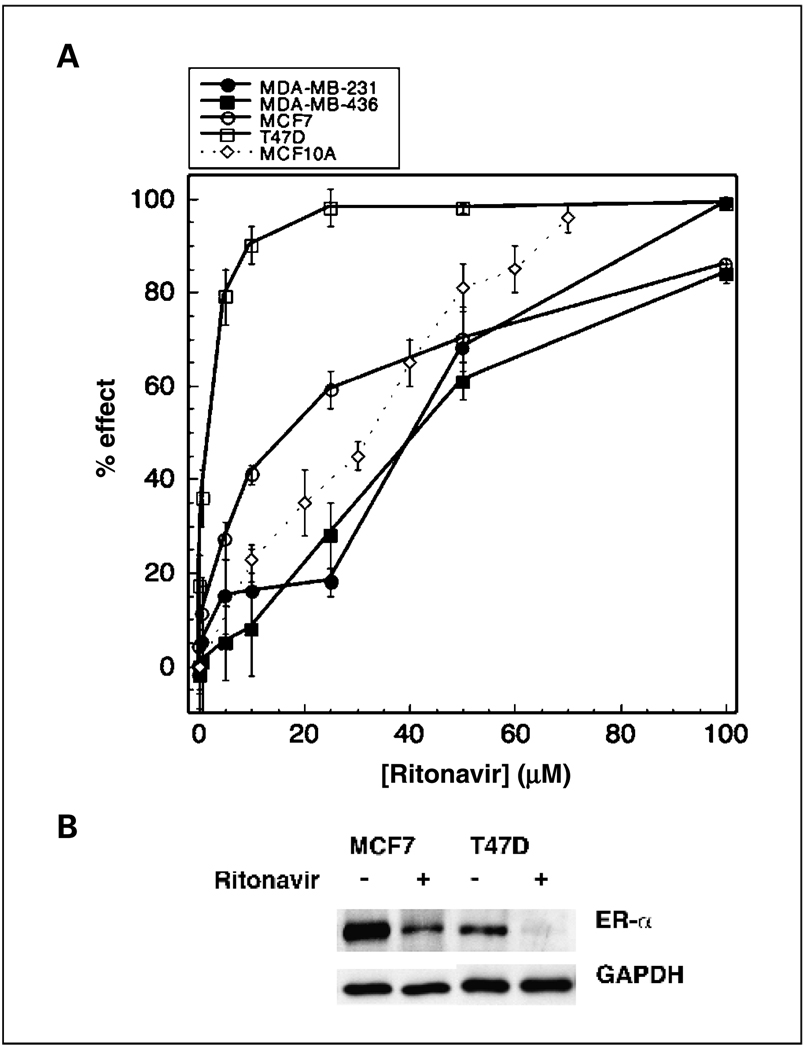
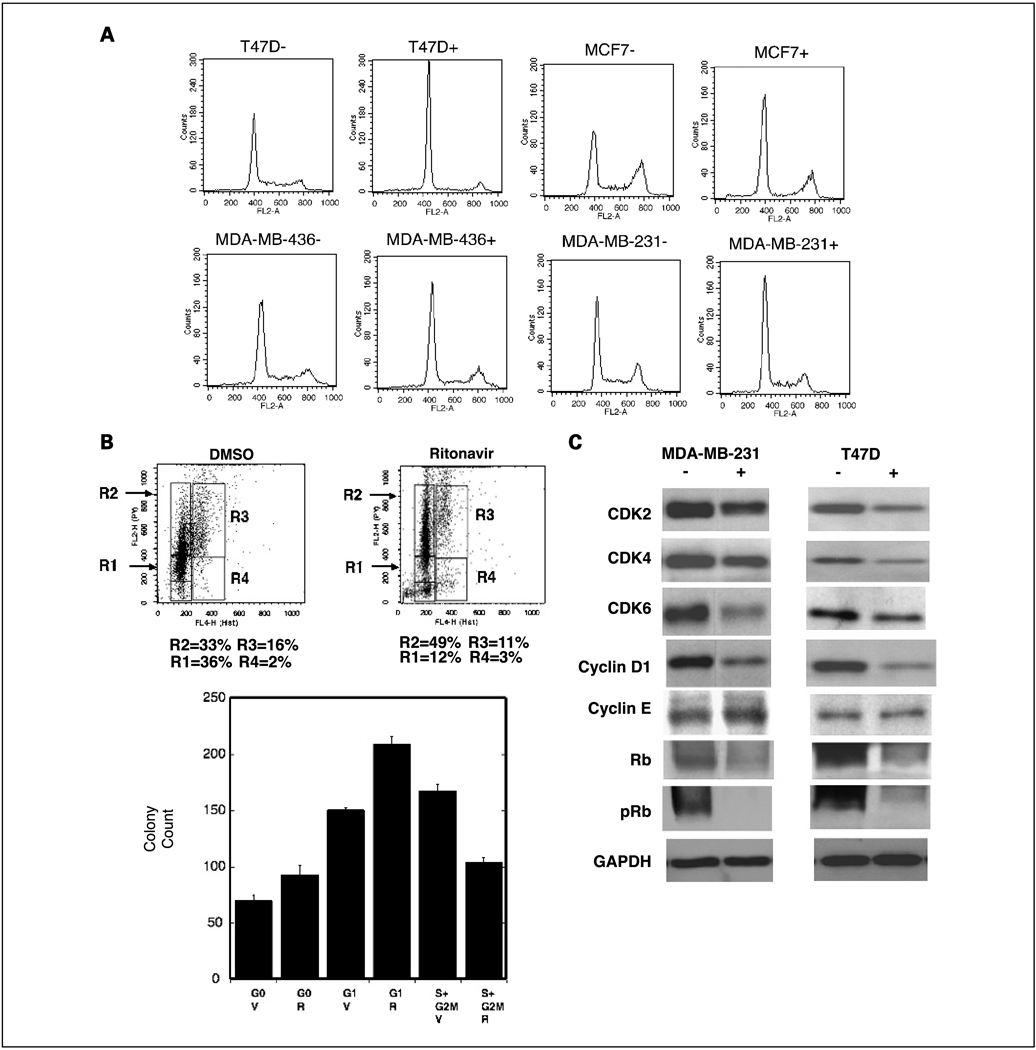
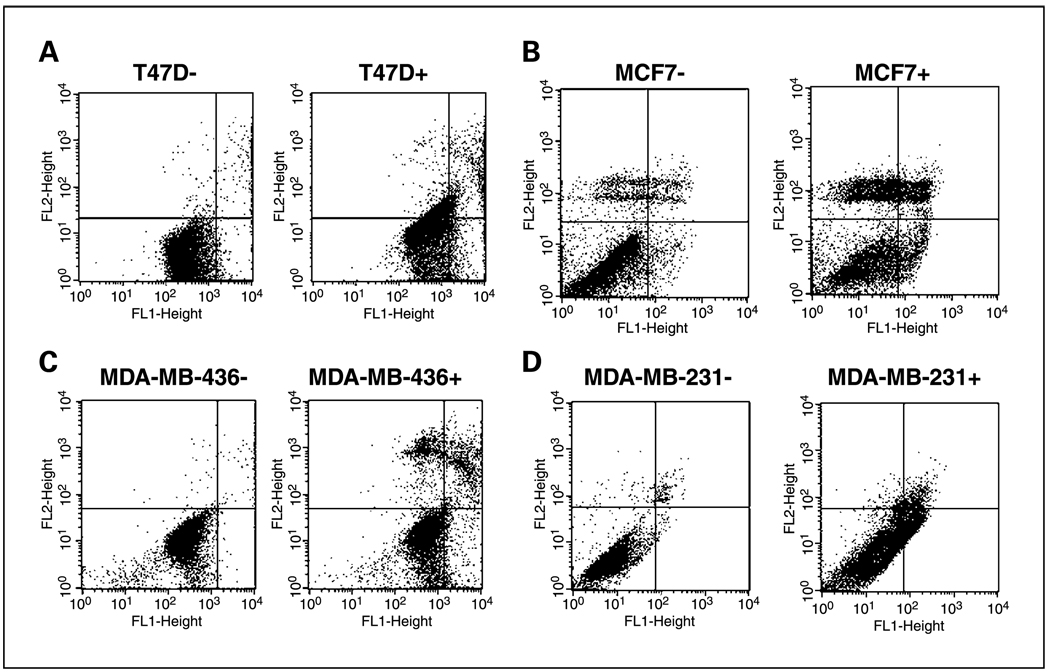
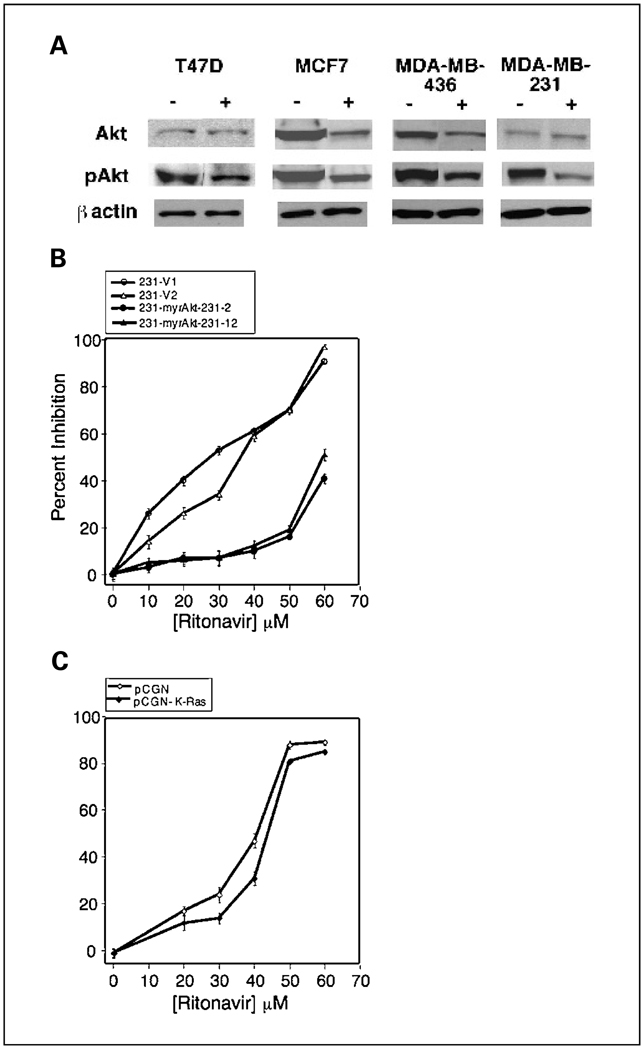
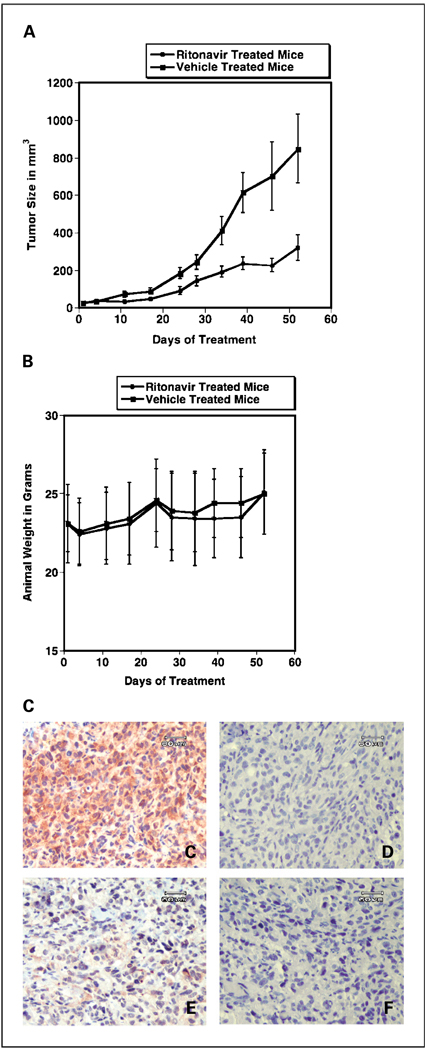
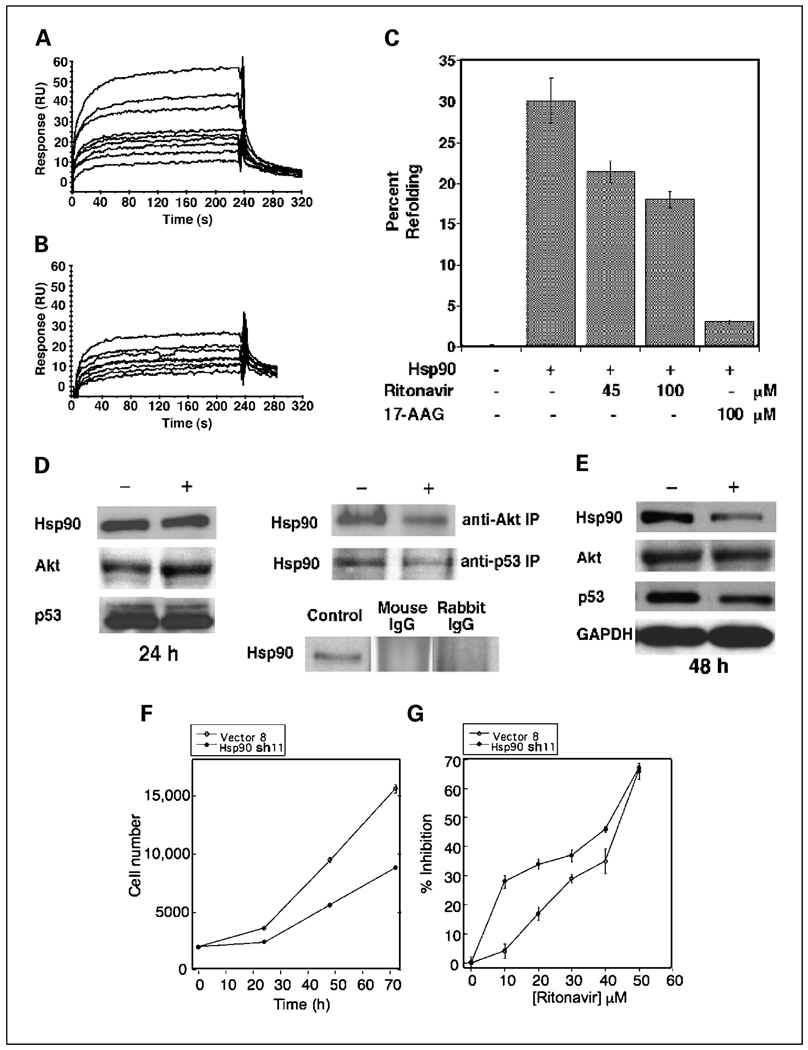
Results: ER-positive estradiol-dependent lines (IC50, 12-24 micromol/L) and ER-negative (IC50, 45 micromol/L) lines exhibit ritonavir sensitivity. Ritonavir depletes ER-alpha levels notably in ER-positive lines. Ritonavir causes G1 arrest, depletes cyclin-dependent kinases 2, 4, and 6 and cyclin D1 but not cyclin E, and depletes phosphorylated Rb and Ser473 Akt. Ritonavir induces apoptosis independent of G1 arrest, inhibiting growth of cells that have passed the G1 checkpoint. Myristoyl-Akt, but not activated K-Ras, rescues ritonavir inhibition. Ritonavir inhibited a MDA-MB-231 xenograft and intratumoral Akt activity at a clinically attainable serum Cmax of 22 +/- 8 micromol/L. Because heat shock protein 90 (Hsp90) substrates are depleted by ritonavir, ritonavir effects on Hsp90 were tested. Ritonavir binds Hsp90 (K(D), 7.8 micromol/L) and partially inhibits its chaperone function. Ritonavir blocks association of Hsp90 with Akt and, with sustained exposure, notably depletes Hsp90. Stably expressed Hsp90alpha short hairpin RNA also depletes Hsp90, inhibiting proliferation and sensitizing breast cancer cells to low ritonavir concentrations.
Conclusions: Ritonavir inhibits breast cancer growth in part by inhibiting Hsp90 substrates, including Akt. Ritonavir may be of interest for breast cancer therapeutics and its efficacy may be increased by sustained exposure or Hsp90 RNA interference.
Figures
Similar articles
-
Effective targeting of triple-negative breast cancer cells by PF-4942847, a novel oral inhibitor of Hsp 90.Clin Cancer Res. 2011 Aug 15;17(16):5432-42. doi: 10.1158/1078-0432.CCR-11-0592. Epub 2011 Jun 29. Clin Cancer Res. 2011. Retraction in: Clin Cancer Res. 2017 Jan 15;23(2):612. doi: 10.1158/1078-0432.CCR-16-2872. PMID: 21715568 Retracted.
-
Capsaicin causes cell-cycle arrest and apoptosis in ER-positive and -negative breast cancer cells by modulating the EGFR/HER-2 pathway.Oncogene. 2010 Jan 14;29(2):285-96. doi: 10.1038/onc.2009.335. Epub 2009 Oct 26. Oncogene. 2010. PMID: 19855437
-
Inhibition of heat shock protein 90 function by 17-allylamino-17-demethoxy-geldanamycin in Hodgkin's lymphoma cells down-regulates Akt kinase, dephosphorylates extracellular signal-regulated kinase, and induces cell cycle arrest and cell death.Clin Cancer Res. 2006 Jan 15;12(2):584-90. doi: 10.1158/1078-0432.CCR-05-1194. Clin Cancer Res. 2006. PMID: 16428504
-
Plant-Based HSP90 Inhibitors in Breast Cancer Models: A Systematic Review.Int J Mol Sci. 2024 May 17;25(10):5468. doi: 10.3390/ijms25105468. Int J Mol Sci. 2024. PMID: 38791506 Free PMC article. Review.
-
Molecular chaperones in mammary cancer growth and breast tumor therapy.J Cell Biochem. 2012 Apr;113(4):1096-103. doi: 10.1002/jcb.23461. J Cell Biochem. 2012. PMID: 22105880 Free PMC article. Review.
Cited by
-
Reproductive toxicity of ritonavir in male: Insight into mouse sperm capacitation.Reprod Toxicol. 2022 Dec;114:1-6. doi: 10.1016/j.reprotox.2022.09.008. Epub 2022 Oct 2. Reprod Toxicol. 2022. PMID: 36198369 Free PMC article.
-
Ritonavir and ixazomib kill bladder cancer cells by causing ubiquitinated protein accumulation.Cancer Sci. 2017 Jun;108(6):1194-1202. doi: 10.1111/cas.13242. Epub 2017 May 20. Cancer Sci. 2017. PMID: 28342223 Free PMC article.
-
Breast cancer, human immunodeficiency virus and highly active antiretroviral treatment; implications for a high-rate seropositive region.Oncol Rev. 2019 Jan 14;13(1):376. doi: 10.4081/oncol.2019.376. eCollection 2019 Jan 14. Oncol Rev. 2019. PMID: 30713605 Free PMC article.
-
Breast Cancer in the Setting of HIV.Patholog Res Int. 2011;2011:925712. doi: 10.4061/2011/925712. Epub 2011 May 25. Patholog Res Int. 2011. PMID: 21660281 Free PMC article.
-
Ritonavir's Evolving Role: A Journey from Antiretroviral Therapy to Broader Medical Applications.Curr Oncol. 2024 Oct 8;31(10):6032-6049. doi: 10.3390/curroncol31100450. Curr Oncol. 2024. PMID: 39451754 Free PMC article. Review.
References
-
- Sledge GW, Neuberg D, Bernardo P, et al. Phase III trial of doxorubicin, paclitaxel, and the combination of doxorubicin and paclitaxel as front-line chemotherapy for metastatic breast cancer: an intergroup trial (E1193) J Clin Oncol. 2003;21:588–592. - PubMed
-
- Pati S, Pelser CB, Dufraine J, Bryant JL, Reitz MS, Jr., Weichold FF. Antitumorigenic effects of HIV protease inhibitor ritonavir: inhibition of Kaposi sarcoma. Blood. 2002;99:3771–3779. - PubMed
-
- Gaedicke S, Firat-Geier E, Constantiniu O, et al. Anti-tumor effect of the human immunodeficiency virus protease inhibitor ritonavir: induction of tumor-cell apoptosis associated with perturbation of proteasomal proteolysis. Cancer Res. 2002;62:6901–6908. - PubMed
-
- Laurent N, de Bouard S, Guillamo JS, et al. Effects of the proteasome inhibitor ritonavir on glioma growth in vitro and in vivo. Mol Cancer Ther. 2004;3:129–136. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
