

Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities
- PMID: 16395396
- PMCID: PMC1323269
- DOI: 10.1172/JCI27291
Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities
Abstract
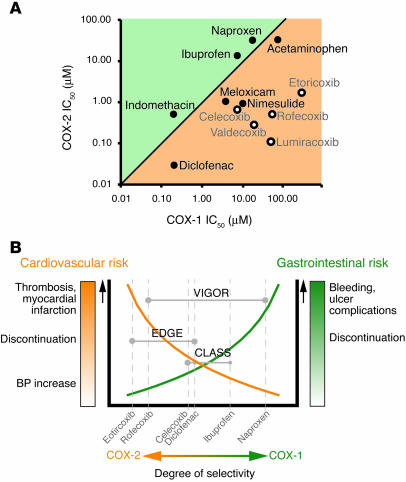
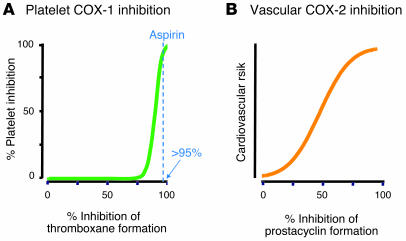
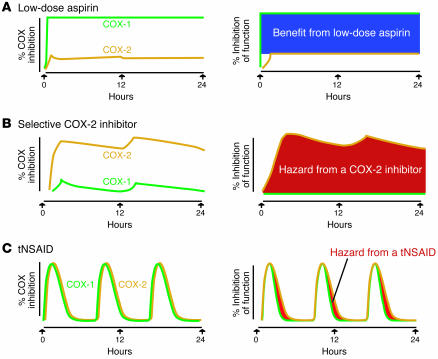
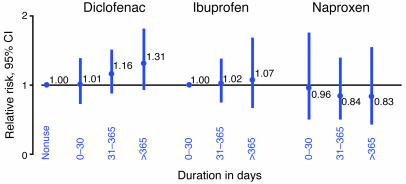
Inhibitors selective for prostaglandin G/H synthase-2 (PGHS-2) (known colloquially as COX-2) were designed to minimize gastrointestinal complications of traditional NSAIDs--adverse effects attributed to suppression of COX-1-derived PGE2 and prostacyclin (PGI2). Evidence from 2 randomized controlled-outcome trials (RCTs) of 2 structurally distinct selective inhibitors of COX-2 supports this hypothesis. However, 5 RCTs of 3 structurally distinct inhibitors also indicate that such compounds elevate the risk of myocardial infarction and stroke. The clinical information is biologically plausible, as it is compatible with evidence that inhibition of COX-2-derived PGI2 removes a protective constraint on thrombogenesis, hypertension, and atherogenesis in vivo. However, the concept of simply tipping a "balance" between COX-2-derived PGI2 and COX-1-derived platelet thromboxane is misplaced. Among the questions that remain to be addressed are the following: (a) whether this hazard extends to all or some of the traditional NSAIDs; (b) whether adjuvant therapies, such as low-dose aspirin, will mitigate the hazard and if so, at what cost; (c) whether COX-2 inhibitors result in cardiovascular risk transformation during chronic dosing; and (d) how we might identify individuals most likely to benefit or suffer from such drugs in the future.
Figures
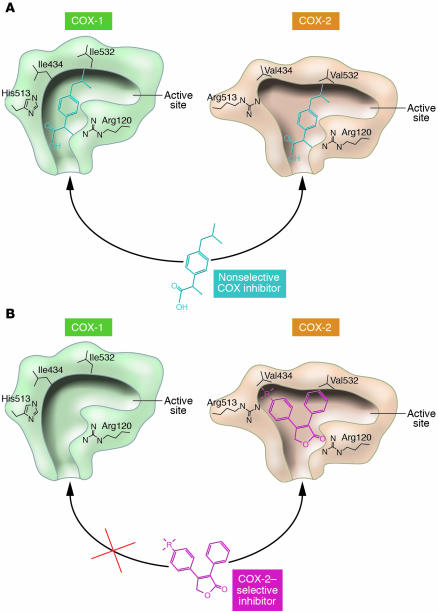
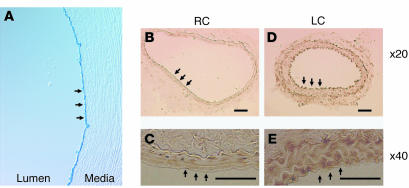
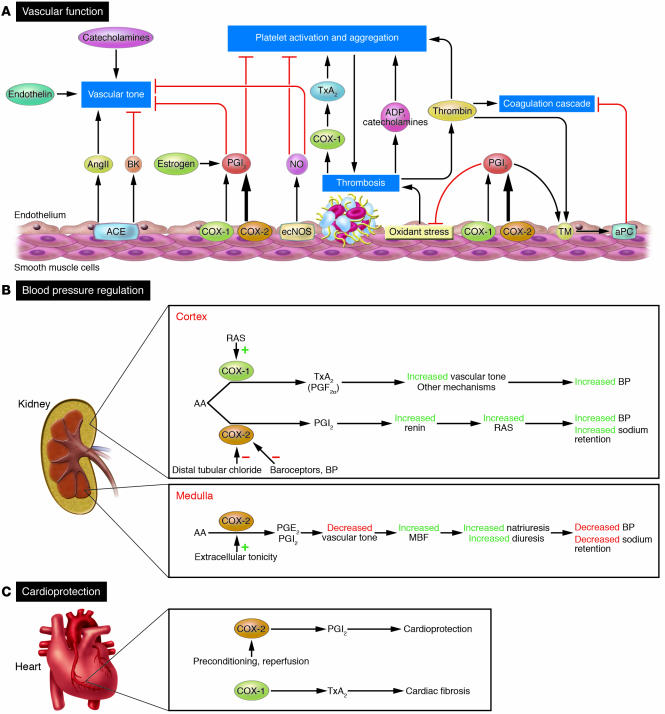
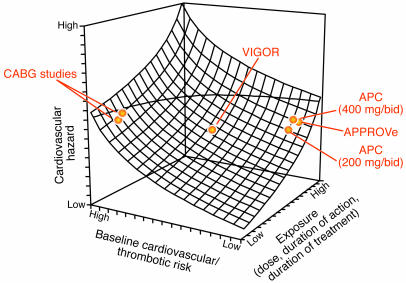
Similar articles
-
The pharmacology of selective inhibition of COX-2.Thromb Haemost. 2006 Oct;96(4):393-400. Thromb Haemost. 2006. PMID: 17003913 Review.
-
The cardiovascular pharmacology of COX-2 inhibition.Hematology Am Soc Hematol Educ Program. 2005:445-51. doi: 10.1182/asheducation-2005.1.445. Hematology Am Soc Hematol Educ Program. 2005. PMID: 16304418
-
Cardiovascular effects of cyclooxygenase-2 inhibitors: a mechanistic and clinical perspective.Br J Clin Pharmacol. 2016 Oct;82(4):957-64. doi: 10.1111/bcp.13048. Epub 2016 Jul 18. Br J Clin Pharmacol. 2016. PMID: 27317138 Free PMC article. Review.
-
Evolution of nonsteroidal anti-inflammatory drugs (NSAIDs): cyclooxygenase (COX) inhibition and beyond.J Pharm Pharm Sci. 2008 Sep 20;11(2):81s-110s. doi: 10.18433/j3t886. J Pharm Pharm Sci. 2008. PMID: 19203472 Review.
-
Cardiovascular complications of non-steroidal anti-inflammatory drugs.Ann Clin Lab Sci. 2005 Autumn;35(4):347-85. Ann Clin Lab Sci. 2005. PMID: 16254252 Review.
Cited by
-
Efficient, large-scale synthesis and preclinical studies of MRS5698, a highly selective A3 adenosine receptor agonist that protects against chronic neuropathic pain.Purinergic Signal. 2015 Sep;11(3):371-87. doi: 10.1007/s11302-015-9459-2. Epub 2015 Jun 27. Purinergic Signal. 2015. PMID: 26111639 Free PMC article.
-
New insights into structural determinants for prostanoid thromboxane A2 receptor- and prostacyclin receptor-G protein coupling.Mol Cell Biol. 2013 Jan;33(2):184-93. doi: 10.1128/MCB.00725-12. Epub 2012 Oct 29. Mol Cell Biol. 2013. PMID: 23109431 Free PMC article.
-
FNTM: a server for predicting functional networks of tissues in mouse.Nucleic Acids Res. 2015 Jul 1;43(W1):W182-7. doi: 10.1093/nar/gkv443. Epub 2015 May 4. Nucleic Acids Res. 2015. PMID: 25940632 Free PMC article.
-
The H2S-releasing naproxen derivative, ATB-346, inhibits alveolar bone loss and inflammation in rats with ligature-induced periodontitis.Med Gas Res. 2015 Feb 27;5:4. doi: 10.1186/s13618-015-0025-3. eCollection 2015. Med Gas Res. 2015. PMID: 25755876 Free PMC article.
-
Full and partial agonists of thromboxane prostanoid receptor unveil fine tuning of receptor superactive conformation and G protein activation.PLoS One. 2013;8(3):e60475. doi: 10.1371/journal.pone.0060475. Epub 2013 Mar 28. PLoS One. 2013. PMID: 23555978 Free PMC article.
References
-
- Smyth, E., Burke, A., and FitzGerald, G.A. 2005. Lipid-derived autacoids. In Goodman & Gilman’s The pharmacological basis of therapeutics. McGraw-Hill. New York, New York, USA. 653–670.
-
- Funk CD. Leukotriene modifiers as potential therapeutics for cardiovascular disease. Nat. Rev. Drug Discov. 2005;4:664–672. - PubMed
-
- Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Annu. Rev. Biochem. 2002;69:145–182. - PubMed
-
- Burke, A., Smyth, E., and FitzGerald, G.A. 2005. Analgesic-anti-pyretic and anti-inflammatory agents and drugs employed in the treatment of gout. In Goodman & Gilman’s The pharmacological basis of therapeutics. McGraw-Hill. New York, New York, USA. 673–715.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
